
Advanced electronic structure methods#
DFT+U#
index:DFT+U
It can be difficult to find the lowest energy solutions with DFT+U. Some strategies for improving this are discussed in cite:PhysRevB.82.195128.
Metal oxide oxidation energies with DFT+U#
We will reconsider here the reaction (see [BROKEN LINK: Metal oxide oxidation energies]) 2 Cu2O + O2 \(\rightleftharpoons\) 4 CuO. We need to compute the energy of each species, now with DFT+U. In cite:PhysRevB.73.195107 they use a U parameter of 4 eV for Cu which gave the best agreement with the experimental value. We will also try that.
Cu2O calculation with U=4.0#
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
i
m
p
o
r
t
A
t
o
m
,
A
t
o
m
s
t
r
y
:
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
C
u
2
O
'
)
.
c
l
o
n
e
(
'
b
u
l
k
/
C
u
2
O
-
U
=
4
.
0
'
)
c
a
l
c
.
s
e
t
(
l
d
a
u
=
T
r
u
e
,
#
t
u
r
n
D
F
T
+
U
o
n
l
d
a
u
t
y
p
e
=
2
,
#
s
e
l
e
c
t
s
i
m
p
l
i
f
i
e
d
r
o
t
a
t
i
o
n
a
l
l
y
i
n
v
a
r
i
a
n
t
o
p
t
i
o
n
l
d
a
u
_
l
u
j
=
{
'
C
u
'
:
{
'
L
'
:
2
,
'
U
'
:
4
.
0
,
'
J
'
:
0
.
0
}
,
'
O
'
:
{
'
L
'
:
-
1
,
'
U
'
:
0
.
0
,
'
J
'
:
0
.
0
}
}
,
l
d
a
u
p
r
i
n
t
=
1
,
i
b
r
i
o
n
=
-
1
,
#
d
o
n
o
t
r
e
r
e
l
a
x
f
o
r
c
e
=
T
r
u
e
,
n
s
w
=
0
)
a
t
o
m
s
=
c
a
l
c
.
l
o
a
d
_
a
t
o
m
s
(
)
p
r
i
n
t
(
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
)
e
x
c
e
p
t
(
F
i
l
e
N
o
t
F
o
u
n
d
E
r
r
o
r
,
V
a
l
u
e
E
r
r
o
r
)
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
e
}
"
)
p
r
i
n
t
(
"
T
h
i
s
e
x
a
m
p
l
e
r
e
q
u
i
r
e
s
a
p
r
e
-
c
o
m
p
u
t
e
d
b
u
l
k
/
C
u
2
O
c
a
l
c
u
l
a
t
i
o
n
.
"
)
-22.32504781
grep -A 3 "LDA+U is selected, type is set to LDAUTYPE" bulk/Cu2O-U=4.0/OUTCAR
LDA+U is selected, type is set to LDAUTYPE = 2
angular momentum for each species LDAUL = 2 -1
U (eV) for each species LDAUU = 4.0 0.0
J (eV) for each species LDAUJ = 0.0 0.0
LDA+U is selected, type is set to LDAUTYPE = 2
angular momentum for each species LDAUL = 2 -1
U (eV) for each species LDAUU = 4.0 0.0
J (eV) for each species LDAUJ = 0.0 0.0
CuO calculation with U=4.0#
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
i
m
p
o
r
t
A
t
o
m
,
A
t
o
m
s
t
r
y
:
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
C
u
O
'
)
c
a
l
c
.
c
l
o
n
e
(
'
b
u
l
k
/
C
u
O
-
U
=
4
.
0
'
)
c
a
l
c
.
s
e
t
(
l
d
a
u
=
T
r
u
e
,
#
t
u
r
n
D
F
T
+
U
o
n
l
d
a
u
t
y
p
e
=
2
,
#
s
e
l
e
c
t
s
i
m
p
l
i
f
i
e
d
r
o
t
a
t
i
o
n
a
l
l
y
i
n
v
a
r
i
a
n
t
o
p
t
i
o
n
l
d
a
u
_
l
u
j
=
{
'
C
u
'
:
{
'
L
'
:
2
,
'
U
'
:
4
.
0
,
'
J
'
:
0
.
0
}
,
'
O
'
:
{
'
L
'
:
-
1
,
'
U
'
:
0
.
0
,
'
J
'
:
0
.
0
}
}
,
l
d
a
u
p
r
i
n
t
=
1
,
i
b
r
i
o
n
=
-
1
,
#
d
o
n
o
t
r
e
r
e
l
a
x
n
s
w
=
0
)
a
t
o
m
s
=
c
a
l
c
.
g
e
t
_
a
t
o
m
s
(
)
p
r
i
n
t
(
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
)
e
x
c
e
p
t
(
F
i
l
e
N
o
t
F
o
u
n
d
E
r
r
o
r
,
V
a
l
u
e
E
r
r
o
r
)
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
e
}
"
)
p
r
i
n
t
(
"
T
h
i
s
e
x
a
m
p
l
e
r
e
q
u
i
r
e
s
a
p
r
e
-
c
o
m
p
u
t
e
d
b
u
l
k
/
C
u
O
c
a
l
c
u
l
a
t
i
o
n
.
"
)
-16.91708676
Reaction energy calculation with DFT+U#
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
t
r
y
:
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
C
u
2
O
-
U
=
4
.
0
'
)
a
t
o
m
s
=
c
a
l
c
.
g
e
t
_
a
t
o
m
s
(
)
c
u
2
o
_
e
n
e
r
g
y
=
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
/
(
l
e
n
(
a
t
o
m
s
)
/
3
)
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
C
u
O
-
U
=
4
.
0
'
)
a
t
o
m
s
=
c
a
l
c
.
g
e
t
_
a
t
o
m
s
(
)
c
u
o
_
e
n
e
r
g
y
=
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
/
(
l
e
n
(
a
t
o
m
s
)
/
2
)
#
m
a
k
e
s
u
r
e
t
o
u
s
e
t
h
e
s
a
m
e
c
u
t
o
f
f
e
n
e
r
g
y
f
o
r
t
h
e
O
2
m
o
l
e
c
u
l
e
!
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
m
o
l
e
c
u
l
e
s
/
O
2
-
s
p
-
t
r
i
p
l
e
t
-
4
0
0
'
)
o
2
_
e
n
e
r
g
y
=
c
a
l
c
.
r
e
s
u
l
t
s
[
'
e
n
e
r
g
y
'
]
c
a
l
c
.
s
t
o
p
_
i
f
(
N
o
n
e
i
n
[
c
u
2
o
_
e
n
e
r
g
y
,
c
u
o
_
e
n
e
r
g
y
,
o
2
_
e
n
e
r
g
y
]
)
#
d
o
n
'
t
f
o
r
g
e
t
t
o
n
o
r
m
a
l
i
z
e
y
o
u
r
t
o
t
a
l
e
n
e
r
g
y
t
o
a
f
o
r
m
u
l
a
u
n
i
t
.
C
u
2
O
#
h
a
s
3
a
t
o
m
s
,
s
o
t
h
e
n
u
m
b
e
r
o
f
f
o
r
m
u
l
a
u
n
i
t
s
i
n
a
n
a
t
o
m
s
i
s
#
l
e
n
(
a
t
o
m
s
)
/
3
.
r
x
n
_
e
n
e
r
g
y
=
4
.
0
*
c
u
o
_
e
n
e
r
g
y
-
o
2
_
e
n
e
r
g
y
-
2
.
0
*
c
u
2
o
_
e
n
e
r
g
y
p
r
i
n
t
(
f
'
R
e
a
c
t
i
o
n
e
n
e
r
g
y
=
{
r
x
n
_
e
n
e
r
g
y
}
e
V
'
)
p
r
i
n
t
(
f
'
C
o
r
r
e
c
t
e
d
e
n
e
r
g
y
=
{
r
x
n
_
e
n
e
r
g
y
-
1
.
3
6
}
e
V
'
)
e
x
c
e
p
t
(
F
i
l
e
N
o
t
F
o
u
n
d
E
r
r
o
r
,
V
a
l
u
e
E
r
r
o
r
)
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
e
}
"
)
p
r
i
n
t
(
"
T
h
i
s
e
x
a
m
p
l
e
r
e
q
u
i
r
e
s
p
r
e
-
c
o
m
p
u
t
e
d
C
u
2
O
,
C
u
O
,
a
n
d
O
2
c
a
l
c
u
l
a
t
i
o
n
s
.
"
)
Reaction energy = 7.36775847 eV
Corrected energy = 6.00775847 eV
This is still not in quantitative agreement with the result in cite:PhysRevB.73.195107, which at U=4 eV is about -3.14 eV (estimated from a graph). We have not applied the O\(_2\) correction here yet. In that paper, they apply a constant shift of -1.36 eV per O\(_2\). After we apply that correction, we agree within 0.12 eV, which is pretty good considering we have not checked for convergence.
How much does U affect the reaction energy?#
It is reasonable to consider how sensitive our results are to the U parameter. We do that here.
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
o
r
U
i
n
[
2
.
0
,
4
.
0
,
6
.
0
]
:
#
#
C
u
2
O
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
t
r
y
:
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
C
u
2
O
'
)
c
a
l
c
.
c
l
o
n
e
(
f
'
b
u
l
k
/
C
u
2
O
-
U
=
{
U
}
'
)
c
a
l
c
.
s
e
t
(
l
d
a
u
=
T
r
u
e
,
#
t
u
r
n
D
F
T
+
U
o
n
l
d
a
u
t
y
p
e
=
2
,
#
s
e
l
e
c
t
s
i
m
p
l
i
f
i
e
d
r
o
t
a
t
i
o
n
a
l
l
y
i
n
v
a
r
i
a
n
t
o
p
t
i
o
n
l
d
a
u
_
l
u
j
=
{
'
C
u
'
:
{
'
L
'
:
2
,
'
U
'
:
U
,
'
J
'
:
0
.
0
}
,
'
O
'
:
{
'
L
'
:
-
1
,
'
U
'
:
0
.
0
,
'
J
'
:
0
.
0
}
}
,
l
d
a
u
p
r
i
n
t
=
1
,
i
b
r
i
o
n
=
-
1
,
#
d
o
n
o
t
r
e
r
e
l
a
x
n
s
w
=
0
)
a
t
o
m
s
1
=
c
a
l
c
.
g
e
t
_
a
t
o
m
s
(
)
c
u
2
o
_
e
n
e
r
g
y
=
a
t
o
m
s
1
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
/
(
l
e
n
(
a
t
o
m
s
1
)
/
3
)
e
x
c
e
p
t
(
F
i
l
e
N
o
t
F
o
u
n
d
E
r
r
o
r
,
V
a
l
u
e
E
r
r
o
r
)
:
p
r
i
n
t
(
f
"
U
=
{
U
}
:
b
u
l
k
/
C
u
2
O
n
o
t
f
o
u
n
d
,
s
k
i
p
p
i
n
g
"
)
c
o
n
t
i
n
u
e
#
#
C
u
O
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
t
r
y
:
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
C
u
O
'
)
c
a
l
c
.
c
l
o
n
e
(
f
'
b
u
l
k
/
C
u
O
-
U
=
{
U
}
'
)
c
a
l
c
.
s
e
t
(
l
d
a
u
=
T
r
u
e
,
#
t
u
r
n
D
F
T
+
U
o
n
l
d
a
u
t
y
p
e
=
2
,
#
s
e
l
e
c
t
s
i
m
p
l
i
f
i
e
d
r
o
t
a
t
i
o
n
a
l
l
y
i
n
v
a
r
i
a
n
t
o
p
t
i
o
n
l
d
a
u
_
l
u
j
=
{
'
C
u
'
:
{
'
L
'
:
2
,
'
U
'
:
U
,
'
J
'
:
0
.
0
}
,
'
O
'
:
{
'
L
'
:
-
1
,
'
U
'
:
0
.
0
,
'
J
'
:
0
.
0
}
}
,
l
d
a
u
p
r
i
n
t
=
1
,
i
b
r
i
o
n
=
-
1
,
#
d
o
n
o
t
r
e
r
e
l
a
x
n
s
w
=
0
)
a
t
o
m
s
2
=
c
a
l
c
.
g
e
t
_
a
t
o
m
s
(
)
c
u
o
_
e
n
e
r
g
y
=
a
t
o
m
s
2
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
/
(
l
e
n
(
a
t
o
m
s
2
)
/
2
)
e
x
c
e
p
t
(
F
i
l
e
N
o
t
F
o
u
n
d
E
r
r
o
r
,
V
a
l
u
e
E
r
r
o
r
)
:
p
r
i
n
t
(
f
"
U
=
{
U
}
:
b
u
l
k
/
C
u
O
n
o
t
f
o
u
n
d
,
s
k
i
p
p
i
n
g
"
)
c
o
n
t
i
n
u
e
#
#
O
2
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
#
m
a
k
e
s
u
r
e
t
o
u
s
e
t
h
e
s
a
m
e
c
u
t
o
f
f
e
n
e
r
g
y
f
o
r
t
h
e
O
2
m
o
l
e
c
u
l
e
!
t
r
y
:
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
m
o
l
e
c
u
l
e
s
/
O
2
-
s
p
-
t
r
i
p
l
e
t
-
4
0
0
'
)
a
t
o
m
s
=
c
a
l
c
.
g
e
t
_
a
t
o
m
s
(
)
o
2
_
e
n
e
r
g
y
=
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
e
x
c
e
p
t
(
F
i
l
e
N
o
t
F
o
u
n
d
E
r
r
o
r
,
V
a
l
u
e
E
r
r
o
r
)
:
p
r
i
n
t
(
f
"
U
=
{
U
}
:
m
o
l
e
c
u
l
e
s
/
O
2
-
s
p
-
t
r
i
p
l
e
t
-
4
0
0
n
o
t
f
o
u
n
d
,
s
k
i
p
p
i
n
g
"
)
c
o
n
t
i
n
u
e
i
f
N
o
n
e
n
o
t
i
n
[
c
u
2
o
_
e
n
e
r
g
y
,
c
u
o
_
e
n
e
r
g
y
,
o
2
_
e
n
e
r
g
y
]
:
r
x
n
_
e
n
e
r
g
y
=
(
4
.
0
*
c
u
o
_
e
n
e
r
g
y
-
o
2
_
e
n
e
r
g
y
-
2
.
0
*
c
u
2
o
_
e
n
e
r
g
y
)
p
r
i
n
t
(
f
'
U
=
{
U
}
r
e
a
c
t
i
o
n
e
n
e
r
g
y
=
{
r
x
n
_
e
n
e
r
g
y
-
1
.
9
9
}
'
)
U = 2.0 reaction energy = 3.32752349
U = 4.0 reaction energy = 5.37775847
U = 6.0 reaction energy = 5.71849513
U = 2.0 reaction energy = -3.876906
U = 4.0 reaction energy = -3.653819
U = 6.0 reaction energy = -3.397605
In cite:PhysRevB.73.195107, the difference in reaction energy from U=2 eV to U=4 eV was about 0.5 eV (estimated from graph). Here we see a range of 0.48 eV from U=2 eV to U=4 eV. Note that for U=0 eV, we had a (corrected reaction energy of -3.96 eV). Overall, the effect of adding U decreases this reaction energy.
This example highlights the challenge of using an approach like DFT+U. On one hand, U has a clear effect of changing the reaction energy. On the other hand, so does the correction factor for the O\(_2\) binding energy. In cite:PhysRevB.73.195107 the authors tried to get the O\(_2\) binding energy correction from oxide calculations where U is not important, so that it is decoupled from the non-cancelling errors that U fixes. See cite:PhysRevB.84.045115 for additional discussion of how to mix GGA and GGA+U results.
In any case, you should be careful to use well converged results to avoid compensating for convergence errors with U.
Hybrid functionals#
FCC Ni DOS#
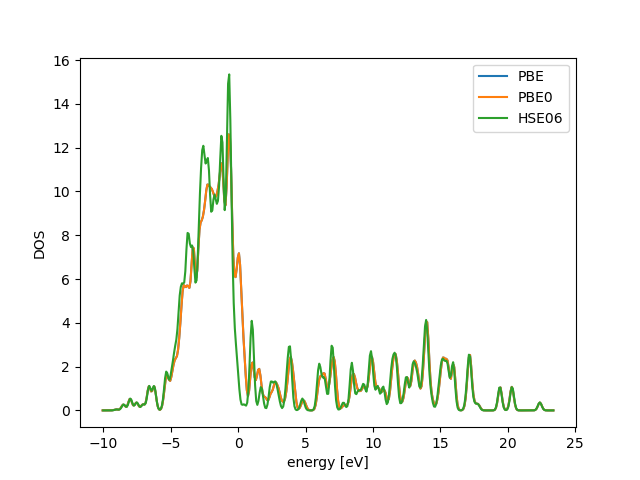
This example is adapted from http://cms.mpi.univie.ac.at/wiki/index.php/FccNi_DOS [BROKEN LINK: index:HSE06]
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
f
c
c
1
1
1
,
b
u
l
k
f
r
o
m
a
s
e
.
d
f
t
i
m
p
o
r
t
D
O
S
i
m
p
o
r
t
m
a
t
p
l
o
t
l
i
b
.
p
y
p
l
o
t
a
s
p
l
t
a
t
o
m
s
=
b
u
l
k
(
'
N
i
'
,
'
f
c
c
'
,
a
=
3
.
5
2
,
c
u
b
i
c
=
T
r
u
e
)
a
t
o
m
s
[
0
]
.
m
a
g
m
o
m
=
1
t
r
y
:
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
N
i
-
P
B
E
'
,
i
s
m
e
a
r
=
-
5
,
k
p
t
s
=
[
5
,
5
,
5
]
,
x
c
=
'
P
B
E
'
,
i
s
p
i
n
=
2
,
l
o
r
b
i
t
=
1
1
,
l
w
a
v
e
=
T
r
u
e
,
l
c
h
a
r
g
=
T
r
u
e
,
a
t
o
m
s
=
a
t
o
m
s
)
e
=
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
p
r
i
n
t
(
'
P
B
E
e
n
e
r
g
y
:
'
,
e
)
i
f
e
i
s
N
o
n
e
:
r
a
i
s
e
V
a
l
u
e
E
r
r
o
r
(
"
P
B
E
e
n
e
r
g
y
n
o
t
a
v
a
i
l
a
b
l
e
"
)
d
o
s
=
D
O
S
(
c
a
l
c
,
w
i
d
t
h
=
0
.
2
)
e
_
p
b
e
=
d
o
s
.
g
e
t
_
e
n
e
r
g
i
e
s
(
)
d
_
p
b
e
=
d
o
s
.
g
e
t
_
d
o
s
(
)
c
a
l
c
.
c
l
o
n
e
(
'
b
u
l
k
/
N
i
-
P
B
E
0
'
)
c
a
l
c
.
s
e
t
(
x
c
=
'
p
b
e
0
'
)
a
t
o
m
s
=
c
a
l
c
.
g
e
t
_
a
t
o
m
s
(
)
p
b
e
0
_
e
=
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
i
f
p
b
e
0
_
e
i
s
n
o
t
N
o
n
e
:
d
o
s
=
D
O
S
(
c
a
l
c
,
w
i
d
t
h
=
0
.
2
)
e
_
p
b
e
0
=
d
o
s
.
g
e
t
_
e
n
e
r
g
i
e
s
(
)
d
_
p
b
e
0
=
d
o
s
.
g
e
t
_
d
o
s
(
)
#
#
H
S
E
0
6
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
N
i
-
P
B
E
'
)
c
a
l
c
.
c
l
o
n
e
(
'
b
u
l
k
/
N
i
-
H
S
E
0
6
'
)
c
a
l
c
.
s
e
t
(
x
c
=
'
h
s
e
0
6
'
)
a
t
o
m
s
=
c
a
l
c
.
g
e
t
_
a
t
o
m
s
(
)
h
s
e
0
6
_
e
=
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
i
f
h
s
e
0
6
_
e
i
s
n
o
t
N
o
n
e
:
d
o
s
=
D
O
S
(
c
a
l
c
,
w
i
d
t
h
=
0
.
2
)
e
_
h
s
e
0
6
=
d
o
s
.
g
e
t
_
e
n
e
r
g
i
e
s
(
)
d
_
h
s
e
0
6
=
d
o
s
.
g
e
t
_
d
o
s
(
)
i
f
N
o
n
e
n
o
t
i
n
[
e
,
p
b
e
0
_
e
,
h
s
e
0
6
_
e
]
:
p
l
t
.
p
l
o
t
(
e
_
p
b
e
,
d
_
p
b
e
,
l
a
b
e
l
=
'
P
B
E
'
)
p
l
t
.
p
l
o
t
(
e
_
p
b
e
0
,
d
_
p
b
e
0
,
l
a
b
e
l
=
'
P
B
E
0
'
)
p
l
t
.
p
l
o
t
(
e
_
h
s
e
0
6
,
d
_
h
s
e
0
6
,
l
a
b
e
l
=
'
H
S
E
0
6
'
)
p
l
t
.
x
l
a
b
e
l
(
'
e
n
e
r
g
y
[
e
V
]
'
)
p
l
t
.
y
l
a
b
e
l
(
'
D
O
S
'
)
p
l
t
.
l
e
g
e
n
d
(
)
p
l
t
.
s
a
v
e
f
i
g
(
'
i
m
a
g
e
s
/
n
i
-
d
o
s
-
p
b
e
-
p
b
e
0
-
h
s
e
0
6
.
p
n
g
'
)
e
x
c
e
p
t
(
F
i
l
e
N
o
t
F
o
u
n
d
E
r
r
o
r
,
V
a
l
u
e
E
r
r
o
r
)
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
e
}
"
)
p
r
i
n
t
(
"
T
h
i
s
e
x
a
m
p
l
e
r
e
q
u
i
r
e
s
b
u
l
k
/
N
i
-
P
B
E
c
a
l
c
u
l
a
t
i
o
n
.
"
)
van der Waals forces#
Older versions (5.2.11+) implement DFT+D2 cite:JCC-JCC20495 with the incar:LVDW tag.
The vdW-DF cite:klimes-2011-van-waals is accessed with incar:LUSEVDW. See http://cms.mpi.univie.ac.at/vasp/vasp/vdW_DF_functional_Langreth_Lundqvist_et_al.htmlfor notes on its usage.
In Vasp 5.3+, the incar:IVDW tag turns van der Waal calculations on.
You should review the links below before using these
IVDW |
method |
|---|---|
0 |
no correction |
1 or 10 |
DFT-D2method of Grimme (available as of VASP.5.2.11) |
11 |
zero damping DFT-D3 method of Grimme (available as of VASP.5.3.4) |
12 |
DFT-D3method with Becke-Jonson damping (available as of VASP.5.3.4) |
2 or 20 |
Tkatchenko-Scheffler methodcite:tkatchenko-2009-accur-molec (available as of VASP.5.3.3) |
Van der Waal forces can play a considerable role in binding of aromatic molecules to metal surfaces (ref). Here we consider the effects of these forces on the adsorption energy of benzene on an Au(111) surface.First, we consider the regular PBE functional.
PBE#
gas-phase benzene#
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
m
o
l
e
c
u
l
e
b
e
n
z
e
n
e
=
m
o
l
e
c
u
l
e
(
'
C
6
H
6
'
)
b
e
n
z
e
n
e
.
c
e
n
t
e
r
(
v
a
c
u
u
m
=
5
)
t
r
y
:
p
r
i
n
t
(
V
a
s
p
(
l
a
b
e
l
=
'
m
o
l
e
c
u
l
e
s
/
b
e
n
z
e
n
e
-
p
b
e
'
,
x
c
=
'
P
B
E
'
,
e
n
c
u
t
=
3
5
0
,
k
p
t
s
=
[
1
,
1
,
1
]
,
i
b
r
i
o
n
=
1
,
n
s
w
=
1
0
0
,
a
t
o
m
s
=
b
e
n
z
e
n
e
)
.
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-76.03718564
clean slab#
#
t
h
e
c
l
e
a
n
g
o
l
d
s
l
a
b
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
f
c
c
1
1
1
,
a
d
d
_
a
d
s
o
r
b
a
t
e
f
r
o
m
a
s
e
.
c
o
n
s
t
r
a
i
n
t
s
i
m
p
o
r
t
F
i
x
A
t
o
m
s
a
t
o
m
s
=
f
c
c
1
1
1
(
'
A
u
'
,
s
i
z
e
=
(
3
,
3
,
3
)
,
v
a
c
u
u
m
=
1
0
)
#
n
o
w
w
e
c
o
n
s
t
r
a
i
n
t
h
e
s
l
a
b
c
=
F
i
x
A
t
o
m
s
(
m
a
s
k
=
[
a
t
o
m
.
s
y
m
b
o
l
=
=
'
A
u
'
f
o
r
a
t
o
m
i
n
a
t
o
m
s
]
)
a
t
o
m
s
.
s
e
t
_
c
o
n
s
t
r
a
i
n
t
(
c
)
t
r
y
:
p
r
i
n
t
(
V
a
s
p
(
l
a
b
e
l
=
'
s
u
r
f
a
c
e
s
/
A
u
-
p
b
e
'
,
x
c
=
'
P
B
E
'
,
e
n
c
u
t
=
3
5
0
,
k
p
t
s
=
[
4
,
4
,
1
]
,
i
b
r
i
o
n
=
1
,
n
s
w
=
1
0
0
,
a
t
o
m
s
=
a
t
o
m
s
)
.
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-81.22521492
benzene on Au(111)#
#
B
e
n
z
e
n
e
o
n
t
h
e
s
l
a
b
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
f
c
c
1
1
1
,
a
d
d
_
a
d
s
o
r
b
a
t
e
,
m
o
l
e
c
u
l
e
f
r
o
m
a
s
e
.
c
o
n
s
t
r
a
i
n
t
s
i
m
p
o
r
t
F
i
x
A
t
o
m
s
a
t
o
m
s
=
f
c
c
1
1
1
(
'
A
u
'
,
s
i
z
e
=
(
3
,
3
,
3
)
,
v
a
c
u
u
m
=
1
0
)
b
e
n
z
e
n
e
=
m
o
l
e
c
u
l
e
(
'
C
6
H
6
'
)
b
e
n
z
e
n
e
.
t
r
a
n
s
l
a
t
e
(
-
b
e
n
z
e
n
e
.
g
e
t
_
c
e
n
t
e
r
_
o
f
_
m
a
s
s
(
)
)
#
I
w
a
n
t
t
h
e
b
e
n
z
e
n
e
c
e
n
t
e
r
e
d
o
n
t
h
e
p
o
s
i
t
i
o
n
i
n
t
h
e
m
i
d
d
l
e
o
f
a
t
o
m
s
#
2
0
,
2
2
,
2
3
a
n
d
2
5
p
=
(
a
t
o
m
s
.
p
o
s
i
t
i
o
n
s
[
2
0
]
+
a
t
o
m
s
.
p
o
s
i
t
i
o
n
s
[
2
2
]
+
a
t
o
m
s
.
p
o
s
i
t
i
o
n
s
[
2
3
]
+
a
t
o
m
s
.
p
o
s
i
t
i
o
n
s
[
2
5
]
)
/
4
.
0
+
[
0
.
0
,
0
.
0
,
3
.
0
5
]
b
e
n
z
e
n
e
.
t
r
a
n
s
l
a
t
e
(
p
)
a
t
o
m
s
+
=
b
e
n
z
e
n
e
#
n
o
w
w
e
c
o
n
s
t
r
a
i
n
t
h
e
s
l
a
b
c
=
F
i
x
A
t
o
m
s
(
m
a
s
k
=
[
a
t
o
m
.
s
y
m
b
o
l
=
=
'
A
u
'
f
o
r
a
t
o
m
i
n
a
t
o
m
s
]
)
a
t
o
m
s
.
s
e
t
_
c
o
n
s
t
r
a
i
n
t
(
c
)
t
r
y
:
p
r
i
n
t
(
V
a
s
p
(
l
a
b
e
l
=
'
s
u
r
f
a
c
e
s
/
A
u
-
b
e
n
z
e
n
e
-
p
b
e
'
,
x
c
=
'
P
B
E
'
,
e
n
c
u
t
=
3
5
0
,
k
p
t
s
=
[
4
,
4
,
1
]
,
i
b
r
i
o
n
=
1
,
n
s
w
=
1
0
0
,
a
t
o
m
s
=
a
t
o
m
s
)
.
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
/home-research/jkitchin/dft-book/surfaces/Au-benzene-pbe submitted: 1413525.gilgamesh.cheme.cmu.edu
None
resubmitted
/home-research/jkitchin/dft-book/surfaces/Au-benzene-pbe submitted: 1399668.gilgamesh.cheme.cmu.edu
None
try:
from vasp import Vasp
e1, e2, e3 = [Vasp(wd).potential_energy
for wd in ['surfaces/Au-benzene-pbe',
'surfaces/Au-pbe',
'molecules/benzene-pbe']]
print('PBE adsorption energy = {} eV'.format(e1 - e2 - e3))
except Exception as e:
print(f"Calculation not available: {type(e).__name__}: {e}")
This is a very weak energy. It is similar to the result in the reference (0.15 eV), and considerably weaker than the experiment. Next we consider one form of a VDW correction.
DFT-D2#
To turn on the van der Waals corrections cite:JCC-JCC20495 we set incar:LVDW to True.
gas-phase benzene#
t
r
y
:
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
m
o
l
e
c
u
l
e
b
e
n
z
e
n
e
=
m
o
l
e
c
u
l
e
(
'
C
6
H
6
'
)
b
e
n
z
e
n
e
.
c
e
n
t
e
r
(
v
a
c
u
u
m
=
5
)
p
r
i
n
t
(
V
a
s
p
(
l
a
b
e
l
=
'
m
o
l
e
c
u
l
e
s
/
b
e
n
z
e
n
e
-
p
b
e
-
d
2
'
,
x
c
=
'
P
B
E
'
,
e
n
c
u
t
=
3
5
0
,
k
p
t
s
=
[
1
,
1
,
1
]
,
i
b
r
i
o
n
=
1
,
n
s
w
=
1
0
0
,
l
v
d
w
=
T
r
u
e
,
a
t
o
m
s
=
b
e
n
z
e
n
e
)
.
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-76.17670701
clean slab#
#
t
h
e
c
l
e
a
n
g
o
l
d
s
l
a
b
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
f
c
c
1
1
1
,
a
d
d
_
a
d
s
o
r
b
a
t
e
f
r
o
m
a
s
e
.
c
o
n
s
t
r
a
i
n
t
s
i
m
p
o
r
t
F
i
x
A
t
o
m
s
a
t
o
m
s
=
f
c
c
1
1
1
(
'
A
u
'
,
s
i
z
e
=
(
3
,
3
,
3
)
,
v
a
c
u
u
m
=
1
0
)
#
n
o
w
w
e
c
o
n
s
t
r
a
i
n
t
h
e
s
l
a
b
c
=
F
i
x
A
t
o
m
s
(
m
a
s
k
=
[
a
t
o
m
.
s
y
m
b
o
l
=
=
'
A
u
'
f
o
r
a
t
o
m
i
n
a
t
o
m
s
]
)
a
t
o
m
s
.
s
e
t
_
c
o
n
s
t
r
a
i
n
t
(
c
)
t
r
y
:
p
r
i
n
t
(
V
a
s
p
(
l
a
b
e
l
=
'
s
u
r
f
a
c
e
s
/
A
u
-
p
b
e
-
d
2
'
,
x
c
=
'
P
B
E
'
,
e
n
c
u
t
=
3
5
0
,
k
p
t
s
=
[
4
,
4
,
1
]
,
i
b
r
i
o
n
=
1
,
n
s
w
=
1
0
0
,
l
v
d
w
=
T
r
u
e
,
a
t
o
m
s
=
a
t
o
m
s
)
.
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-106.34723065
benzene on Au(111)#
t
r
y
:
#
B
e
n
z
e
n
e
o
n
t
h
e
s
l
a
b
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
f
c
c
1
1
1
,
a
d
d
_
a
d
s
o
r
b
a
t
e
,
m
o
l
e
c
u
l
e
f
r
o
m
a
s
e
.
c
o
n
s
t
r
a
i
n
t
s
i
m
p
o
r
t
F
i
x
A
t
o
m
s
a
t
o
m
s
=
f
c
c
1
1
1
(
'
A
u
'
,
s
i
z
e
=
(
3
,
3
,
3
)
,
v
a
c
u
u
m
=
1
0
)
b
e
n
z
e
n
e
=
m
o
l
e
c
u
l
e
(
'
C
6
H
6
'
)
b
e
n
z
e
n
e
.
t
r
a
n
s
l
a
t
e
(
-
b
e
n
z
e
n
e
.
g
e
t
_
c
e
n
t
e
r
_
o
f
_
m
a
s
s
(
)
)
#
I
w
a
n
t
t
h
e
b
e
n
z
e
n
e
c
e
n
t
e
r
e
d
o
n
t
h
e
p
o
s
i
t
i
o
n
i
n
t
h
e
m
i
d
d
l
e
o
f
a
t
o
m
s
#
2
0
,
2
2
,
2
3
a
n
d
2
5
p
=
(
a
t
o
m
s
.
p
o
s
i
t
i
o
n
s
[
2
0
]
+
a
t
o
m
s
.
p
o
s
i
t
i
o
n
s
[
2
2
]
+
a
t
o
m
s
.
p
o
s
i
t
i
o
n
s
[
2
3
]
+
a
t
o
m
s
.
p
o
s
i
t
i
o
n
s
[
2
5
]
)
/
4
.
0
+
[
0
.
0
,
0
.
0
,
3
.
0
5
]
b
e
n
z
e
n
e
.
t
r
a
n
s
l
a
t
e
(
p
)
a
t
o
m
s
+
=
b
e
n
z
e
n
e
#
n
o
w
w
e
c
o
n
s
t
r
a
i
n
t
h
e
s
l
a
b
c
=
F
i
x
A
t
o
m
s
(
m
a
s
k
=
[
a
t
o
m
.
s
y
m
b
o
l
=
=
'
A
u
'
f
o
r
a
t
o
m
i
n
a
t
o
m
s
]
)
a
t
o
m
s
.
s
e
t
_
c
o
n
s
t
r
a
i
n
t
(
c
)
#
f
r
o
m
a
s
e
.
v
i
s
u
a
l
i
z
e
i
m
p
o
r
t
v
i
e
w
;
v
i
e
w
(
a
t
o
m
s
)
p
r
i
n
t
(
V
a
s
p
(
l
a
b
e
l
=
'
s
u
r
f
a
c
e
s
/
A
u
-
b
e
n
z
e
n
e
-
p
b
e
-
d
2
'
,
x
c
=
'
P
B
E
'
,
e
n
c
u
t
=
3
5
0
,
k
p
t
s
=
[
4
,
4
,
1
]
,
i
b
r
i
o
n
=
1
,
n
s
w
=
1
0
0
,
l
v
d
w
=
T
r
u
e
,
a
t
o
m
s
=
a
t
o
m
s
)
.
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-184.07495285
try:
from vasp import Vasp
e1, e2, e3 = [Vasp(wd).potential_energy
for wd in ['surfaces/Au-benzene-pbe-d2',
'surfaces/Au-pbe-d2',
'molecules/benzene-pbe-d2']]
print('Adsorption energy = {0:1.2f} eV'.format(e1 - e2 - e3))
except Exception as e:
print(f"Calculation not available: {type(e).__name__}: {e}")
Adsorption energy = -1.54 eV
Adsorption energy = -1.54 eV
That is significantly more favorable. This is much higher than this reference though (0.56 eV), so there could be some issues with convergence or other computational parameters that should be considered.
Electron localization function#
The electron localization function (ELF) can be used to characterize chemical bonds, e.g. their ionicity/covalency cite:silvi1994. Here we reproduce an example from Ref. citenum:silvi1994. We compute and plot the ELF for tetrafluoromethane. The incar:LELF tag turns this on.
t
r
y
:
#
c
o
m
p
u
t
e
E
L
F
f
o
r
C
F
4
#
N
o
t
e
:
T
h
i
s
c
e
l
l
r
e
q
u
i
r
e
s
e
n
t
h
o
u
g
h
t
.
m
a
y
a
v
i
f
o
r
3
D
v
i
s
u
a
l
i
z
a
t
i
o
n
#
w
h
i
c
h
i
s
n
o
t
a
v
a
i
l
a
b
l
e
i
n
t
h
i
s
e
n
v
i
r
o
n
m
e
n
t
.
T
h
e
c
a
l
c
u
l
a
t
i
o
n
s
e
t
u
p
i
s
s
h
o
w
n
b
e
l
o
w
.
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
m
o
l
e
c
u
l
e
a
t
o
m
s
=
m
o
l
e
c
u
l
e
(
'
C
F
4
'
)
a
t
o
m
s
.
c
e
n
t
e
r
(
v
a
c
u
u
m
=
5
)
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
m
o
l
e
c
u
l
e
s
/
c
f
4
-
e
l
f
'
,
e
n
c
u
t
=
3
5
0
,
p
r
e
c
=
'
h
i
g
h
'
,
i
s
m
e
a
r
=
0
,
s
i
g
m
a
=
0
.
0
1
,
x
c
=
'
P
B
E
'
,
l
e
l
f
=
T
r
u
e
,
a
t
o
m
s
=
a
t
o
m
s
)
p
r
i
n
t
(
"
C
F
4
E
L
F
c
a
l
c
u
l
a
t
i
o
n
s
e
t
u
p
c
o
m
p
l
e
t
e
"
)
p
r
i
n
t
(
"
T
o
v
i
s
u
a
l
i
z
e
E
L
F
,
u
s
e
m
a
y
a
v
i
:
m
l
a
b
.
c
o
n
t
o
u
r
3
d
(
x
,
y
,
z
,
e
l
f
,
c
o
n
t
o
u
r
s
=
[
0
.
3
]
)
"
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
None


These images (Figure ref:fig:elf1 and ref:fig:elf2) are basically consistent with those in Reference cite:silvi1994.
Charge partitioning schemes#
Modeling Core level shifts#
We need to setup four calculations. First, we setup the bulk Cu and bulk alloy calculations and let them relax. We use similar unit cells for each one to maximize cancellation of errors.
t
r
y
:
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
i
m
p
o
r
t
A
t
o
m
,
A
t
o
m
s
a
t
o
m
s
=
A
t
o
m
s
(
[
A
t
o
m
(
'
C
u
'
,
[
0
.
0
0
0
,
0
.
0
0
0
,
0
.
0
0
0
]
)
,
A
t
o
m
(
'
C
u
'
,
[
-
1
.
6
5
2
,
0
.
0
0
0
,
2
.
0
3
9
]
)
]
,
c
e
l
l
=
[
[
0
.
0
0
0
,
-
2
.
0
3
9
,
2
.
0
3
9
]
,
[
0
.
0
0
0
,
2
.
0
3
9
,
2
.
0
3
9
]
,
[
-
3
.
3
0
3
,
0
.
0
0
0
,
0
.
0
0
0
]
]
)
a
t
o
m
s
=
a
t
o
m
s
.
r
e
p
e
a
t
(
(
2
,
2
,
2
)
)
p
r
i
n
t
(
a
t
o
m
s
[
0
]
)
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
C
u
-
c
l
s
-
0
'
,
x
c
=
'
P
B
E
'
,
e
n
c
u
t
=
3
5
0
,
k
p
t
s
=
[
4
,
4
,
4
]
,
i
b
r
i
o
n
=
2
,
i
s
i
f
=
3
,
n
s
w
=
4
0
,
a
t
o
m
s
=
a
t
o
m
s
)
p
r
i
n
t
(
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
Atom('Cu', [0.0, 0.0, 0.0], index=0)
-59.98232341
Here, we setup the alloy calculation.
try:
from vasp import Vasp
from ase import Atom, Atoms
atoms = Atoms([Atom('Cu', [0.000, 0.000, 0.000]),
Atom('Pd', [-1.652, 0.000, 2.039])],
cell= [[0.000, -2.039, 2.039],
[0.000, 2.039, 2.039],
[-3.303, 0.000, 0.000]])
atoms = atoms.repeat((2, 2, 2))
calc = Vasp(label='bulk/CuPd-cls-0',
xc='PBE',
encut=350,
kpts=(4, 4, 4),
ibrion=2,
isif=3,
nsw=40,
atoms=atoms)
print(atoms.get_potential_energy())
except Exception as e:
print(f"Calculation not available: {type(e).__name__}: {e}")
-73.55012322
-73.55012322
Next, we have to do the excitation in each structure. For these, we do not relax the structure. We clone the previous results and modify them.
t
r
y
:
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
b
u
l
k
/
C
u
-
c
l
s
-
0
'
)
c
a
l
c
.
c
l
o
n
e
(
'
b
u
l
k
/
C
u
-
c
l
s
-
1
'
)
c
a
l
c
.
s
e
t
(
i
b
r
i
o
n
=
N
o
n
e
,
i
s
i
f
=
N
o
n
e
,
n
s
w
=
N
o
n
e
,
s
e
t
u
p
s
=
[
[
0
,
'
C
u
'
]
]
,
#
C
r
e
a
t
e
s
e
p
a
r
a
t
e
e
n
t
r
y
i
n
P
O
T
C
A
R
f
o
r
a
t
o
m
i
n
d
e
x
0
i
c
o
r
e
l
e
v
e
l
=
2
,
#
P
e
r
f
o
r
m
c
o
r
e
l
e
v
e
l
s
h
i
f
t
c
a
l
c
u
l
a
t
i
o
n
c
l
n
t
=
0
,
#
E
x
c
i
t
e
a
t
o
m
i
n
d
e
x
0
c
l
n
=
2
,
#
2
p
3
/
2
e
l
e
c
t
r
o
n
f
o
r
C
u
c
o
r
e
l
e
v
e
l
s
h
i
f
t
c
l
l
=
1
,
c
l
z
=
1
)
c
a
l
c
.
u
p
d
a
t
e
(
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-345.05440951
try:
from vasp import Vasp
calc = Vasp(label='bulk/CuPd-cls-0')
calc.clone('bulk/CuPd-cls-1')
calc.set(ibrion=None,
isif=None,
nsw=None,
setups=[[0, 'Cu']], # Create separate entry in POTCAR for atom index 0
icorelevel=2, # Perform core level shift calculation
clnt=0, # Excite atom index 0
cln=2, # 2p3/2 electron for Cu core level shift
cll=1,
clz=1)
calc.update()
except Exception as e:
print(f"Calculation not available: {type(e).__name__}: {e}")
-359.87250408
-359.87250408
Finally we calculate the CLS:
try:
from vasp import Vasp
alloy_0 = Vasp(label='bulk/CuPd-cls-0').potential_energy
alloy_1 = Vasp(label='bulk/CuPd-cls-1').potential_energy
ref_0 = Vasp(label='bulk/Cu-cls-0').potential_energy
ref_1 = Vasp(label='bulk/Cu-cls-1').potential_energy
CLS = (alloy_1 - alloy_0) - (ref_1 - ref_0)
print('CLS = {} eV'.format(CLS))
except Exception as e:
print(f"Calculation not available: {type(e).__name__}: {e}")
CLS = -1.2378242 eV
CLS = -1.2378242 eV
This is a little negative compared to the literature but that could be due to the highly ordered structure we used.
The BEEF functional in Vasp#
In Vasp 5.3.5 it is possible to use the BEEF functional cite:wellendorff-2012-densit.
some addtional variables to setup van der Waals and to get the BEEF ensemble energies. Let us consider the dissociation energy of H2.
t
r
y
:
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
m
o
l
e
c
u
l
e
i
m
p
o
r
t
m
a
t
p
l
o
t
l
i
b
.
p
y
p
l
o
t
a
s
p
l
t
H
2
=
m
o
l
e
c
u
l
e
(
'
H
2
'
)
H
2
.
s
e
t
_
c
e
l
l
(
[
8
,
8
,
8
]
,
s
c
a
l
e
_
a
t
o
m
s
=
F
a
l
s
e
)
H
2
.
c
e
n
t
e
r
(
)
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
m
o
l
e
c
u
l
e
s
/
H
2
-
b
e
e
f
'
,
x
c
=
'
b
e
e
f
-
v
d
w
'
,
e
n
c
u
t
=
3
5
0
,
i
s
m
e
a
r
=
0
,
i
b
r
i
o
n
=
2
,
n
s
w
=
1
0
,
a
t
o
m
s
=
H
2
)
e
H
2
=
H
2
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
p
r
i
n
t
(
e
H
2
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-7.13332059
Next, we get an H atom.
t
r
y
:
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
m
o
l
e
c
u
l
e
H
=
m
o
l
e
c
u
l
e
(
'
H
'
)
H
.
s
e
t
_
c
e
l
l
(
[
8
,
8
,
8
]
,
s
c
a
l
e
_
a
t
o
m
s
=
F
a
l
s
e
)
H
.
c
e
n
t
e
r
(
)
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
m
o
l
e
c
u
l
e
s
/
H
-
b
e
e
f
'
,
x
c
=
'
b
e
e
f
-
v
d
w
'
,
e
n
c
u
t
=
3
5
0
,
i
s
m
e
a
r
=
0
,
a
t
o
m
s
=
H
)
p
r
i
n
t
(
c
a
l
c
.
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-0.22476997
Now, the dissociation energy.
try:
from vasp import Vasp
print('D = {} eV'.format(2 * Vasp(label='molecules/H-beef').potential_energy -
Vasp(label='molecules/H2-beef').potential_energy))
except Exception as e:
print(f"Calculation not available: {type(e).__name__}: {e}")
D = 6.68378065 eV
D = 6.68378065 eV
-1.15994056 -7.13332059
D = 4.81343947 eV
It doesn’t look like we have done much so far. How certain are we of the dissociation energy? Let us consider the ensemble of energies. In the calculation, an ensemble of functionals is used, and each one produces a different energy. We can look at the distribution of these energies to estimate the uncertainty in energy differences. We use the func:Vasp.getbeefens to get the ensemble. We calculate the uncertainty in our reaction energy by calculating the standard deviation of the appropriately weighted difference of ensembles.
Note that this ensemble represents the contribution just from the functionals, and not all the other contributions. So, the differences in the ensembles only represents that part of the uncertainty
try:
from vasp import Vasp
calc = Vasp(label='molecules/H-beef')
ensH = calc.get_beefens()
calc = Vasp(label='molecules/H2-beef')
ensH2 = calc.get_beefens()
ensD = 2 * ensH - ensH2
print('mean = {} eV'.format(ensD.mean()))
print('std = {} eV'.format(ensD.std()))
import matplotlib.pyplot as plt
plt.hist(ensD, 20)
plt.xlabel('Deviation')
plt.ylabel('frequency')
except Exception as e:
print(f"Calculation not available: {type(e).__name__}: {e}")
mean = 0.00661973433552 eV
std = 0.278495927893 eV
mean = 0.00661973433552 eV
std = 0.278495927893 eV
You can see the mean is nearly zero, suggesting the deviations are symmetrically distributed. The std error is 0.184 eV, which represents about a 68% confidence interval.

Solvation#
See http://vaspsol.mse.ufl.edu/download/, cite:fishman-2013-accur,mathew-2014-implic
You need a specially patched version of Vasp.
First, we run our calculation in vacuum. We need this to get the WAVECAR. The following calculation mimics one of the example calculations in the Vaspsol package. The combination of nsw=0 and ibrion=2 does not make sense, but that is the example. I do not use the npar=4 parameter here.
t
r
y
:
f
r
o
m
v
a
s
p
i
m
p
o
r
t
V
a
s
p
f
r
o
m
a
s
e
.
b
u
i
l
d
i
m
p
o
r
t
m
o
l
e
c
u
l
e
a
t
o
m
s
=
m
o
l
e
c
u
l
e
(
'
C
O
'
)
a
t
o
m
s
.
c
e
n
t
e
r
(
v
a
c
u
u
m
=
5
)
c
a
l
c
=
V
a
s
p
(
l
a
b
e
l
=
'
m
o
l
e
c
u
l
e
s
/
C
O
-
v
a
c
u
u
m
'
,
e
n
c
u
t
=
6
0
0
,
p
r
e
c
=
'
A
c
c
u
r
a
t
e
'
,
i
s
m
e
a
r
=
0
,
s
i
g
m
a
=
0
.
0
5
,
i
b
r
i
o
n
=
2
,
n
s
w
=
0
,
e
d
i
f
f
=
1
e
-
6
,
a
t
o
m
s
=
a
t
o
m
s
)
p
r
i
n
t
(
a
t
o
m
s
.
g
e
t
_
p
o
t
e
n
t
i
a
l
_
e
n
e
r
g
y
(
)
)
p
r
i
n
t
(
a
t
o
m
s
.
g
e
t
_
f
o
r
c
e
s
(
)
)
p
r
i
n
t
(
f
'
C
a
l
c
u
l
a
t
i
o
n
t
i
m
e
:
{
c
a
l
c
.
g
e
t
_
e
l
a
p
s
e
d
_
t
i
m
e
(
)
}
s
e
c
o
n
d
s
'
)
e
x
c
e
p
t
E
x
c
e
p
t
i
o
n
a
s
e
:
p
r
i
n
t
(
f
"
C
a
l
c
u
l
a
t
i
o
n
n
o
t
a
v
a
i
l
a
b
l
e
:
{
t
y
p
e
(
e
)
.
_
_
n
a
m
e
_
_
}
:
{
e
}
"
)
-14.81547852
[[ 0. 0. -0.949]
[ 0. 0. 0.949]]
Calculation time: 257.546 seconds
The forces are high because nsw was set to 0, so only one iteration was run.
Next, we do the solvation calculation. We use the default solvent dielectric constant of water, which is 80.
try:
from vasp import Vasp
calc = Vasp(label='molecules/CO-vacuum')
calc.clone('molecules/CO-solvated')
calc.set(istart=1, #
lsol=True)
print(calc.get_atoms().get_potential_energy())
print(calc.get_atoms().get_forces())
print('Calculation time: {} seconds'.format(calc.get_elapsed_time()))
except Exception as e:
print(f"Calculation not available: {type(e).__name__}: {e}")
-14.82289079
[[ 0. 0. -1.007]
[ 0. 0. 1.007]]
Calculation time: 2937.72 seconds
-14.82289079
[[ 0. 0. -1.007]
[ 0. 0. 1.007]]
Calculation time: 2937.72 seconds
Note these take quite a bit longer to calculate (e.g. 10 times longer)! The energies here are a little different than the vacuum result. To use this energy in an energy difference, you need to make sure the other energies were run with lsol=True also, and the same parameters.
Here is the evidence that we actually ran a calculation with solvation:
grep -A 5 Solvation molecules/CO-solvated/OUTCAR
LSOL = T Solvation
Electronic Relaxation 1
ENCUT = 600.0 eV 44.10 Ry 6.64 a.u. 19.97 19.97 22.27*2*pi/ulx,y,z
ENINI = 600.0 initial cutoff
ENAUG = 644.9 eV augmentation charge cutoff
--
Solvation parameters
EB_K = 80.000000 relative permittivity of the bulk solvent
SIGMA_K = 0.600000 width of the dielectric cavity
NC_K = 0.002500 cutoff charge density
TAU = 0.000525 cavity surface tension
--
Solvation contrib. Ediel = -2.06361062
---------------------------------------------------
free energy TOTEN = -14.82417510 eV
energy without entropy = -14.82417510 energy(sigma->0) = -14.82417510
--
Solvation contrib. Ediel = -2.08692034
---------------------------------------------------
free energy TOTEN = -14.82331872 eV
energy without entropy = -14.82331872 energy(sigma->0) = -14.82331872
--
Solvation contrib. Ediel = -2.11316669
---------------------------------------------------
free energy TOTEN = -14.82319429 eV
energy without entropy = -14.82319429 energy(sigma->0) = -14.82319429
--
Solvation contrib. Ediel = -2.16318931
---------------------------------------------------
free energy TOTEN = -14.82278947 eV
energy without entropy = -14.82278947 energy(sigma->0) = -14.82278947
--
Solvation contrib. Ediel = -2.17570687
---------------------------------------------------
free energy TOTEN = -14.82272160 eV
energy without entropy = -14.82272160 energy(sigma->0) = -14.82272160
--
Solvation contrib. Ediel = -2.19188585
---------------------------------------------------
free energy TOTEN = -14.82267271 eV
energy without entropy = -14.82267271 energy(sigma->0) = -14.82267271
--
Solvation contrib. Ediel = -2.19395757
---------------------------------------------------
free energy TOTEN = -14.82272442 eV
energy without entropy = -14.82272442 energy(sigma->0) = -14.82272442
--
Solvation contrib. Ediel = -2.19698448
---------------------------------------------------
free energy TOTEN = -14.82288242 eV
energy without entropy = -14.82288242 energy(sigma->0) = -14.82288242
--
Solvation contrib. Ediel = -2.19737905
---------------------------------------------------
free energy TOTEN = -14.82288470 eV
energy without entropy = -14.82288470 energy(sigma->0) = -14.82288470
--
Solvation contrib. Ediel = -2.19908571
---------------------------------------------------
free energy TOTEN = -14.82287091 eV
energy without entropy = -14.82287091 energy(sigma->0) = -14.82287091
--
Solvation contrib. Ediel = -2.19782575
---------------------------------------------------
free energy TOTEN = -14.82288497 eV
energy without entropy = -14.82288497 energy(sigma->0) = -14.82288497
--
Solvation contrib. Ediel = -2.19878993
---------------------------------------------------
free energy TOTEN = -14.82288031 eV
energy without entropy = -14.82288031 energy(sigma->0) = -14.82288031
--
Solvation contrib. Ediel = -2.19875585
---------------------------------------------------
free energy TOTEN = -14.82288727 eV
energy without entropy = -14.82288727 energy(sigma->0) = -14.82288727
--
Solvation contrib. Ediel = -2.19894718
---------------------------------------------------
free energy TOTEN = -14.82288935 eV
energy without entropy = -14.82288935 energy(sigma->0) = -14.82288935
--
Solvation contrib. Ediel = -2.19902584
---------------------------------------------------
free energy TOTEN = -14.82289064 eV
energy without entropy = -14.82289064 energy(sigma->0) = -14.82289064
--
Solvation contrib. Ediel = -2.19905589
---------------------------------------------------
free energy TOTEN = -14.82289079 eV
energy without entropy = -14.82289079 energy(sigma->0) = -14.82289079
LSOL = T Solvation
Electronic Relaxation 1
ENCUT = 600.0 eV 44.10 Ry 6.64 a.u. 19.97 19.97 22.27*2*pi/ulx,y,z
ENINI = 600.0 initial cutoff
ENAUG = 644.9 eV augmentation charge cutoff
--
Solvation parameters
EB_K = 80.000000 relative permittivity of the bulk solvent
SIGMA_K = 0.600000 width of the dielectric cavity
NC_K = 0.002500 cutoff charge density
TAU = 0.000525 cavity surface tension
--
Solvation contrib. Ediel = -2.06361062
---------------------------------------------------
free energy TOTEN = -14.82417510 eV
energy without entropy = -14.82417510 energy(sigma->0) = -14.82417510
--
Solvation contrib. Ediel = -2.08692034
---------------------------------------------------
free energy TOTEN = -14.82331872 eV
energy without entropy = -14.82331872 energy(sigma->0) = -14.82331872
--
Solvation contrib. Ediel = -2.11316669
---------------------------------------------------
free energy TOTEN = -14.82319429 eV
energy without entropy = -14.82319429 energy(sigma->0) = -14.82319429
--
Solvation contrib. Ediel = -2.16318931
---------------------------------------------------
free energy TOTEN = -14.82278947 eV
energy without entropy = -14.82278947 energy(sigma->0) = -14.82278947
--
Solvation contrib. Ediel = -2.17570687
---------------------------------------------------
free energy TOTEN = -14.82272160 eV
energy without entropy = -14.82272160 energy(sigma->0) = -14.82272160
--
Solvation contrib. Ediel = -2.19188585
---------------------------------------------------
free energy TOTEN = -14.82267271 eV
energy without entropy = -14.82267271 energy(sigma->0) = -14.82267271
--
Solvation contrib. Ediel = -2.19395757
---------------------------------------------------
free energy TOTEN = -14.82272442 eV
energy without entropy = -14.82272442 energy(sigma->0) = -14.82272442
--
Solvation contrib. Ediel = -2.19698448
---------------------------------------------------
free energy TOTEN = -14.82288242 eV
energy without entropy = -14.82288242 energy(sigma->0) = -14.82288242
--
Solvation contrib. Ediel = -2.19737905
---------------------------------------------------
free energy TOTEN = -14.82288470 eV
energy without entropy = -14.82288470 energy(sigma->0) = -14.82288470
--
Solvation contrib. Ediel = -2.19908571
---------------------------------------------------
free energy TOTEN = -14.82287091 eV
energy without entropy = -14.82287091 energy(sigma->0) = -14.82287091
--
Solvation contrib. Ediel = -2.19782575
---------------------------------------------------
free energy TOTEN = -14.82288497 eV
energy without entropy = -14.82288497 energy(sigma->0) = -14.82288497
--
Solvation contrib. Ediel = -2.19878993
---------------------------------------------------
free energy TOTEN = -14.82288031 eV
energy without entropy = -14.82288031 energy(sigma->0) = -14.82288031
--
Solvation contrib. Ediel = -2.19875585
---------------------------------------------------
free energy TOTEN = -14.82288727 eV
energy without entropy = -14.82288727 energy(sigma->0) = -14.82288727
--
Solvation contrib. Ediel = -2.19894718
---------------------------------------------------
free energy TOTEN = -14.82288935 eV
energy without entropy = -14.82288935 energy(sigma->0) = -14.82288935
--
Solvation contrib. Ediel = -2.19902584
---------------------------------------------------
free energy TOTEN = -14.82289064 eV
energy without entropy = -14.82289064 energy(sigma->0) = -14.82289064
--
Solvation contrib. Ediel = -2.19905589
---------------------------------------------------
free energy TOTEN = -14.82289079 eV
energy without entropy = -14.82289079 energy(sigma->0) = -14.82289079