Defining and visualizing bulk systems
Defining and visualizing bulk systems
Built-in functions in ase
As with molecules, mod:ase provides several helper functions to create bulk structures. We highlight a few of them here. Particularly common ones are:
mod:ase.lattice.cubic.FaceCenteredCubic
mod:ase.lattice.cubic.BodyCenteredCubic
mod:ase.lattice.hexagonal.Graphite
mod:ase.lattice.compounds.NaCl
For others, see https://wiki.fysik.dtu.dk/ase/ase/lattice.html
We start with a simple example, fcc Ag. By default, mod:ase knows Ag is an fcc metal, and knows the experimental lattice constant. We have to specify the directions (vectors along each axis) to get something other than the default output. Here, the default fcc cell contains four atoms.
Lattice(symbols='Ag4', pbc=True, cell=[4.09, 4.09, 4.09])
A mod:ase.lattice.bravais.Lattice object is returned! This is practically the same as as an mod:ase.atoms.Atoms object.
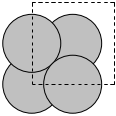
Here we specify the primitive unit cell, which only has one atom in it.
Lattice(symbols='Ag', pbc=True, cell=[[2.892066735052979, 0.0, 0.0], [1.4460333675264898, 2.5046032619957996, 0.0], [1.4460333675264896, 0.8348677539985998, 2.3613626009855695]])
Lattice(symbols='Ag', positions=..., cell=[[2.892066735052979, 0.0, 0.0], [1.4460333675264898, 2.5046032619957996, 0.0], [1.4460333675264896, 0.8348677539985997, 2.3613626009855695]], pbc=[True, True, True])
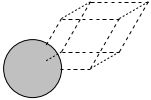
Lattice(symbols='Ag', positions=..., cell=[[2.892066735052979, 0.0, 0.0], [1.4460333675264898, 2.5046032619957996, 0.0], [1.4460333675264896, 0.8348677539985997, 2.3613626009855695]], pbc=[True, True, True])
We can use these modules to build alloy unit cells. The basic strategy is to create the base unit cell in one element and then selectively change some atoms to different chemical symbols. Here we examine an Ag3Pd alloy structure.
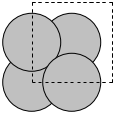
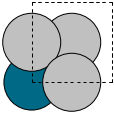
To create a graphite structure we use the following code. Note that we have to specify the lattice constants (taken from http://www.phy.ohiou.edu/~asmith/NewATOMS/HOPG.pdf) because mod:ase has C in the diamond structure by default. We show two views, because the top view does not show the spacing between the layers.
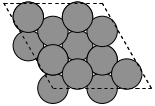
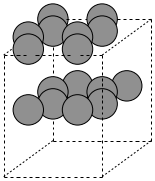
To get a compound, we use the following code. We have to specify the basis atoms to the function generating the compound, and the lattice constant. For NaCl we use the lattice constant at (http://en.wikipedia.org/wiki/Sodium_chloride).
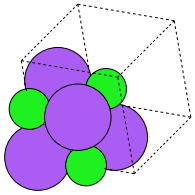
mod:ase.spacegroup
A final alternative to setting up bulk structures is mod:ase.spacegroup. This is a concise way to setup structures if you know the following properties of the crystal structure:
Chemical symbols
Coordinates of the non-equivalent sites in the unit cell
the spacegroup
the cell parameters (a, b, c, alpha, beta, gamma)
Atoms(symbols='Al4', pbc=True, cell=[4.05, 4.05, 4.05], spacegroup_kinds=...)
Atoms(symbols='Al4', positions=..., cell=[[4.05, 0.0, 0.0], [2.4799097682733903e-16, 4.05, 0.0], [2.4799097682733903e-16, 2.4799097682733903e-16, 4.05]], pbc=[True, True, True])
Here is rutile TiO2.
Atoms(symbols='Ti2O4', pbc=True, cell=[4.6, 4.6, 2.95], spacegroup_kinds=...)
Atoms(symbols’Ti2O4’, positions=…, cell=[[4.6, 0.0, 0.0], [2.816687638038912e-16, 4.6, 0.0], [1.806354028742346e-16, 1.806354028742346e-16, 2.95]], pbc=[True, True, True])
=Atoms(symbols’Ti2O4’, positions=…, cell=[[4.6, 0.0, 0.0], [2.816687638038912e-16, 4.6, 0.0], [1.806354028742346e-16, 1.806354028742346e-16, 2.95]], pbc=[True, True, True])
=sho
The Materials Project offers web access to a pretty large number of materials (over 21,000 at the time of this writing), including structure and other computed properties. You must sign up for an account at the website, and then you can access the information. You can search for materials with lots of different criteria including formula, unit cell formula, by elements, by structure, etc… The website allows you to download the VASP files used to create the calculations. They also develop the pymatgen project (which requires python 2.7+).
For example, I downloaded this cif file for a RuO\(_2\) structure (Material ID 825).
#\#CIF1.1
##########################################################################
# Crystallographic Information Format file
# Produced by PyCifRW module
#
# This is a CIF file. CIF has been adopted by the International
# Union of Crystallography as the standard for data archiving and
# transmission.
#
# For information on this file format, follow the CIF links at
# http://www.iucr.org
##########################################################################
data_RuO2
_symmetry_space_group_name_H-M 'P 1'
_cell_length_a 3.13970109
_cell_length_b 4.5436378
_cell_length_c 4.5436378
_cell_angle_alpha 90.0
_cell_angle_beta 90.0
_cell_angle_gamma 90.0
_chemical_name_systematic 'Generated by pymatgen'
_symmetry_Int_Tables_number 1
_chemical_formula_structural RuO2
_chemical_formula_sum 'Ru2 O4'
_cell_volume 64.8180127062
_cell_formula_units_Z 2
loop_
_symmetry_equiv_pos_site_id
_symmetry_equiv_pos_as_xyz
1 'x, y, z'
loop_
_atom_site_type_symbol
_atom_site_label
_atom_site_symmetry_multiplicity
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_attached_hydrogens
_atom_site_B_iso_or_equiv
_atom_site_occupancy
O O1 1 0.000000 0.694330 0.694330 0 . 1
O O2 1 0.500000 0.805670 0.194330 0 . 1
O O3 1 0.000000 0.305670 0.305670 0 . 1
O O4 1 0.500000 0.194330 0.805670 0 . 1
Ru Ru5 1 0.500000 0.500000 0.500000 0 . 1
Ru Ru6 1 0.000000 0.000000 0.000000 0 . 1
We can read this file in with func:ase.io.read. That function automatically recognizes the file type by the extension.
Atoms(symbols='O4Ru2', pbc=True, cell=[3.13970109, 4.5436378, 4.5436378], spacegroup_kinds=...)
