
Magnetism#
Determining if a magnetic solution is energetically favorable#
We can force a total magnetic moment onto a unit cell and compute the total energy as function of the total magnetic moment. If there is a minimum in the energy, then we know there is a lower energy magnetic solution than a non-magnetic solution. We use incar:NUPDOWN to enforce the magnetic moment in the cell. Note that NUPDOWN can only be an integer. You cannot set it to be an arbitrary float.
from vasp import Vasp
from ase.lattice.cubic import BodyCenteredCubic
atoms = BodyCenteredCubic(directions=[[1, 0, 0],
[0, 1, 0],
[0, 0, 1]],
size=(1, 1, 1),
symbol='Fe')
calc = Vasp(label=f'bulk/Fe-bcc-fixedmagmom-{0.0:1.2f}',
xc='PBE',
encut=300,
kpts=(4, 4, 4),
ispin=2,
nupdown=0,
atoms=atoms)
print(atoms.get_potential_energy())
-15.34226599
from vasp import Vasp
from ase.lattice.cubic import BodyCenteredCubic
atoms = BodyCenteredCubic(directions=[[1, 0, 0],
[0, 1, 0],
[0, 0, 1]],
size=(1, 1, 1),
symbol='Fe')
NUPDOWNS = [0.0, 2.0, 4.0, 5.0, 6.0, 8.0]
energies = []
for B in NUPDOWNS:
calc = Vasp(label=f'bulk/Fe-bcc-fixedmagmom-{B:1.2f}',
xc='PBE',
encut=300,
kpts=(4, 4, 4),
ispin=2,
nupdown=B,
atoms=atoms)
energies.append(atoms.get_potential_energy())
if None in energies:
calc.abort()
import matplotlib.pyplot as plt
plt.plot(NUPDOWNS, energies)
plt.xlabel('Total Magnetic Moment')
plt.ylabel('Energy (eV)');
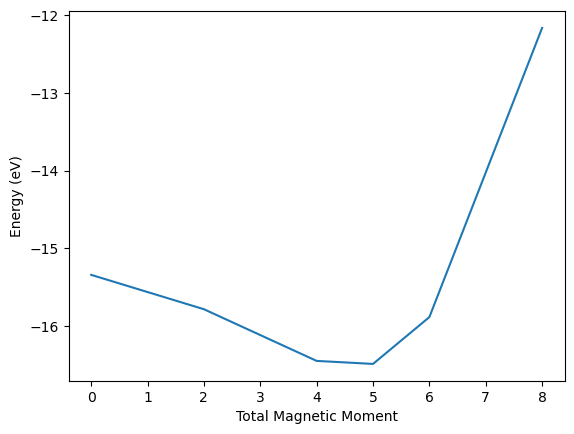
You can see here there is a minimum in energy at a total magnetic moment somewhere between 4 and 5. There are two Fe atoms in the unit cell, which means the magnetic moment on each atom must be about 2.5 Bohr-magnetons. This is a good guess for a real calculation. Note that VASP recommends you overestimate the magnetic moment guesses if you are looking for ferromagnetic solutions.
To run a spin-polarized calculation with initial guesses on each atom, we must set the magnetic moment on the atoms. Here we set it through the magmom attribute on the atom. In the example after this, we set it in the Atoms object.
from vasp import Vasp
from ase.lattice.cubic import BodyCenteredCubic
from ase.calculators.calculator import PropertyNotImplementedError
atoms = BodyCenteredCubic(directions=[[1, 0, 0],
[0, 1, 0],
[0, 0, 1]],
size=(1, 1, 1),
symbol='Fe')
# set magnetic moments on each atom
for atom in atoms:
atom.magmom = 2.5
calc = Vasp(label='bulk/Fe-bcc-sp-1',
xc='PBE',
encut=300,
kpts=(4, 4, 4),
ispin=2,
lorbit=11, # you need this for individual magnetic moments
atoms=atoms)
e = atoms.get_potential_energy()
B = atoms.get_magnetic_moment()
try:
magmoms = atoms.get_magnetic_moments()
print(f'Total magnetic moment is {B:1.2f} Bohr-magnetons')
print(f'Individual moments are {magmoms} Bohr-magnetons')
except PropertyNotImplementedError:
print(f'Total magnetic moment is {B:1.2f} Bohr-magnetons')
print('Note: Individual magnetic moments not available - run VASP with lorbit=11')
Total magnetic moment is 2.00 Bohr-magnetons
Note: Individual magnetic moments not available - run VASP with lorbit=11
Total magnetic moment is -0.01 Bohr-magnetons
Individual moments are [-0.013 -0.013] Bohr-magnetons
Antiferromagnetic spin states#
In an antiferromagnetic material, there are equal numbers of spin up and down electrons that align in a regular pattern, but pointing in opposite directions so that there is no net magnetism. It is possible to model this by setting the magnetic moments on each mod:ase.Atom object. incar:lreal
from vasp import Vasp
from ase import Atom, Atoms
atoms = Atoms([Atom('Fe', [0.00, 0.00, 0.00], magmom=5),
Atom('Fe', [4.3, 4.3, 4.3], magmom=-5),
Atom('O', [2.15, 2.15, 2.15], magmom=0),
Atom('O', [6.45, 6.45, 6.45], magmom=0)],
cell=[[4.3, 2.15, 2.15],
[2.15, 4.3, 2.15],
[2.15, 2.15, 4.3]])
ca = Vasp(label='bulk/afm-feo',
encut=350,
prec='Normal',
ispin=2,
nupdown=0, # this forces a non-magnetic solution
lorbit=11, # to get individual moments
lreal=False,
atoms=atoms)
ca.calculate()
print('Magnetic moments = ', atoms.get_magnetic_moments())
print('Total magnetic moment = ', atoms.get_magnetic_moment())
---------------------------------------------------------------------------
PropertyNotImplementedError Traceback (most recent call last)
Cell In[33], line 23
12 ca = Vasp(label='bulk/afm-feo',
13 encut=350,
14 prec='Normal',
(...)
18 lreal=False,
19 atoms=atoms)
21 ca.calculate()
---> 23 print('Magnetic moments = ', atoms.get_magnetic_moments())
24 print('Total magnetic moment = ', atoms.get_magnetic_moment())
File /opt/conda/lib/python3.9/site-packages/ase/atoms.py:652, in Atoms.get_magnetic_moments(self)
650 if self._calc is None:
651 raise RuntimeError('Atoms object has no calculator.')
--> 652 return self._calc.get_magnetic_moments(self)
File /opt/conda/lib/python3.9/site-packages/ase/calculators/abc.py:45, in GetPropertiesMixin.get_magnetic_moments(self, atoms)
43 def get_magnetic_moments(self, atoms=None):
44 """Calculate magnetic moments projected onto atoms."""
---> 45 return self.get_property('magmoms', atoms)
File /opt/conda/lib/python3.9/site-packages/ase/calculators/calculator.py:742, in Calculator.get_property(self, name, atoms, allow_calculation)
737 self.calculate(atoms, [name], system_changes)
739 if name not in self.results:
740 # For some reason the calculator was not able to do what we want,
741 # and that is OK.
--> 742 raise PropertyNotImplementedError('{} not present in this '
743 'calculation'.format(name))
745 result = self.results[name]
746 if isinstance(result, np.ndarray):
PropertyNotImplementedError: magmoms not present in this calculation
Magnetic moments = [-0.061 -0.061 0.063 0.063]
Total magnetic moment = -5e-06
You can see that even though the total magnetic moment is 0, there is a spin on both Fe atoms, and they are pointing in opposite directions. Hence, the sum of spins is zero, and this arrangement is called anti-ferromagnetic.