Defining and visualizing molecules#
We start by learning how to define a molecule and visualize it. We will begin with defining molecules from scratch, then reading molecules from data files, and finally using some built-in databases in ase.
From scratch#
When there is no data file for the molecule you want, or no database to get it from, you have to define your atoms geometry by hand. Here is how that is done for a CO molecule (Figure ref:fig:co-origin). We must define the type and position of each atom, and the unit cell the atoms are in.
from ase import Atoms, Atom
from ase.io import write
# define an Atoms object
atoms = Atoms([Atom('C', [0., 0., 0.]),
Atom('O', [1.1, 0., 0.])],
cell=(10, 10, 10))
print('V = {0:1.0f} Angstrom^3'.format(atoms.get_volume()))
write('images/simple-cubic-cell.png', atoms, show_unit_cell=2)
V = 1000 Angstrom^3
V = 1000 Angstrom^3
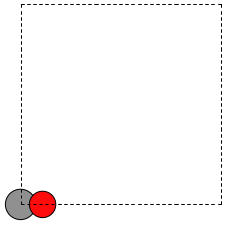
There are two inconvenient features of the simple cubic cell:
Since the CO molecule is at the corner, its electron density is spread over the 8 corners of the box, which is not convenient for visualization later (see [BROKEN LINK: *Visualizing%20electron%20density]).
Due to the geometry of the cube, you need fairly large cubes to make sure the electron density of the molecule does not overlap with that of its images. Electron-electron interactions are repulsive, and the overlap makes the energy increase significantly. Here, the CO molecule has 6 images due to periodic boundary conditions that are 10 Å away. The volume of the unit cell is 1000 Å\(^3\).
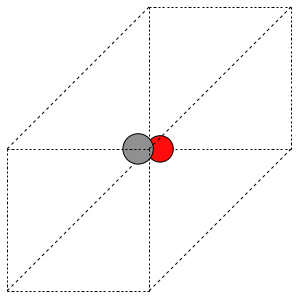
The first problem is easily solved by centering the atoms in the unit cell. The second problem can be solved by using a face-centered cubic lattice, which is the lattice with the closest packing. We show the results of the centering in Figure ref:fig:co-fcc, where we have guessed values for \(b\) until the CO molecules are on average 10 Å apart. Note the final volume is only about 715 Å\(^3\), which is smaller than the cube. This will result in less computational time to compute properties.
from ase import Atoms, Atom
from ase.io import write
b = 7.1
atoms = Atoms([Atom('C', [0., 0., 0.]),
Atom('O', [1.1, 0., 0.])],
cell=[[b, b, 0.],
[b, 0., b],
[0., b, b]])
print('V = {0:1.0f} Ang^3'.format(atoms.get_volume()))
atoms.center() # translate atoms to center of unit cell
write('images/fcc-cell.png', atoms, show_unit_cell=2)
V = 716 Ang^3
V = 716 Ang^3
At this point you might ask, “How do you know the distance to the neighboring image?” The ag viewer lets you compute this graphically, but we can use code to determine this too. All we have to do is figure out the length of each lattice vector, because these are what separate the atoms in the images. We use the mod:numpy module to compute the distance of a vector as the square root of the sum of squared elements.
from ase import Atoms, Atom
import numpy as np
b = 7.1
atoms = Atoms([Atom('C', [0., 0., 0.]),
Atom('O', [1.1, 0., 0.])],
cell=[[b, b, 0.],
[b, 0., b],
[0., b, b]])
# get unit cell vectors and their lengths
(a1, a2, a3) = atoms.get_cell()
print('|a1| = {0:1.2f} Ang'.format(np.sum(a1**2)**0.5))
print('|a2| = {0:1.2f} Ang'.format(np.linalg.norm(a2)))
print('|a3| = {0:1.2f} Ang'.format(np.sum(a3**2)**0.5))
|a1| = 10.04 Ang
|a2| = 10.04 Ang
|a3| = 10.04 Ang
|a1| = 10.04 Ang
|a2| = 10.04 Ang
|a3| = 10.04 Ang
Reading other data formats into a calculation#
mod:ase.io.read supports many different file formats:
Known formats:
========================= ===========
format short name
========================= ===========
GPAW restart-file gpw
Dacapo netCDF output file dacapo
Old ASE netCDF trajectory nc
Virtual Nano Lab file vnl
ASE pickle trajectory traj
ASE bundle trajectory bundle
GPAW text output gpaw-text
CUBE file cube
XCrySDen Structure File xsf
Dacapo text output dacapo-text
XYZ-file xyz
VASP POSCAR/CONTCAR file vasp
VASP OUTCAR file vasp_out
SIESTA STRUCT file struct_out
ABINIT input file abinit
V_Sim ascii file v_sim
Protein Data Bank pdb
CIF-file cif
FHI-aims geometry file aims
FHI-aims output file aims_out
VTK XML Image Data vti
VTK XML Structured Grid vts
VTK XML Unstructured Grid vtu
TURBOMOLE coord file tmol
TURBOMOLE gradient file tmol-gradient
exciting input exi
AtomEye configuration cfg
WIEN2k structure file struct
DftbPlus input file dftb
CASTEP geom file cell
CASTEP output file castep
CASTEP trajectory file geom
ETSF format etsf.nc
DFTBPlus GEN format gen
CMR db/cmr-file db
CMR db/cmr-file cmr
LAMMPS dump file lammps
Gromacs coordinates gro
========================= ===========
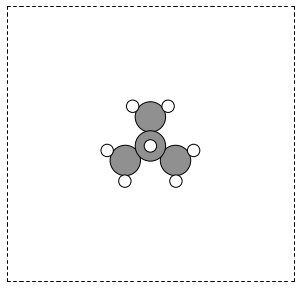
You can read XYZ file format to create mod:ase.Atoms objects. Here is what an XYZ file format might look like:
#+include: molecules/isobutane.xyz
The first line is the number of atoms in the file. The second line is often a comment. What follows is one line per atom with the symbol and Cartesian coordinates in Å. Note that the XYZ format does not have unit cell information in it, so you will have to figure out a way to provide it. In this example, we center the atoms in a box with vacuum on all sides (Figure ref:fig:isobutane).
from ase.io import read, write
atoms = read('molecules/isobutane.xyz')
atoms.center(vacuum=5)
write('images/isobutane-xyz.png', atoms, show_unit_cell=2)
Predefined molecules#
mod:ase defines a number of molecular geometries in the mod:ase.data.molecules database. For example, the database includes the molecules in the G2/97 database cite:curtiss:1063. This database contains a broad set of atoms and molecules for which good experimental data exists, making them useful for benchmarking studies. See this site for the original files.
The coordinates for the atoms in the database are MP2(full)/6-31G(d) optimized geometries. Here is a list of all the species available in mod:ase.data.g2. You may be interested in reading about some of the other databases in mod:ase.data too.
from ase.data import g2
keys = list(g2.data.keys())
# print in 3 columns
for i in range(len(keys) // 3):
print('{0:25s}{1:25s}{2:25s}'.format(*tuple(keys[i * 3: i * 3 + 3])))
Be BeH C
C2H2 C2H4 C2H6
CH CH2_s1A1d CH2_s3B1d
CH3 CH3Cl CH3OH
CH3SH CH4 CN
CO CO2 CS
Cl Cl2 ClF
ClO F F2
H H2CO H2O
H2O2 HCN HCO
HCl HF HOCl
Li Li2 LiF
LiH N N2
N2H4 NH NH2
NH3 NO Na
Na2 NaCl O
O2 OH P
P2 PH2 PH3
S S2 SH2
SO SO2 Si
Si2 Si2H6 SiH2_s1A1d
SiH2_s3B1d SiH3 SiH4
SiO 2-butyne Al
AlCl3 AlF3 B
BCl3 BF3 C2Cl4
C2F4 C2H3 C2H5
C2H6CHOH C2H6NH C2H6SO
C3H4_C2v C3H4_C3v C3H4_D2d
C3H6_Cs C3H6_D3h C3H7
C3H7Cl C3H8 C3H9C
C3H9N C4H4NH C4H4O
C4H4S C5H5N C5H8
C6H6 CCH CCl4
CF3CN CF4 CH2NHCH2
CH2OCH2 CH2SCH2 CH3CH2Cl
CH3CH2NH2 CH3CH2O CH3CH2OCH3
CH3CH2OH CH3CH2SH CH3CHO
CH3CN CH3CO CH3COCH3
CH3COCl CH3COF CH3CONH2
CH3COOH CH3NO2 CH3O
CH3OCH3 CH3ONO CH3S
CH3SCH3 CH3SiH3 COF2
CS2 ClF3 ClNO
F2O H2 H2CCHCN
H2CCHCl H2CCHF H2CCO
H2CCl2 H2CF2 H2COH
H3CNH2 HCCl3 HCF3
HCOOCH3 HCOOH N2O
NCCN NF3 NO2
O3 OCHCHO OCS
PF3 SH SiCl4
SiF4 bicyclobutane butadiene
cyclobutane cyclobutene isobutane
isobutene methylenecyclopropane trans-butane
Some other databases include the mod:ase.data.s22 for weakly interacting dimers and complexes, and mod:ase.data.extramolecules which has a few extras like biphenyl and C60.
Here is an example of getting the geometry of an acetonitrile molecule and writing an image to a file. Note that the default unit cell is a 1 Å × Å × Å cubic cell. That is too small to use if your calculator uses periodic boundary conditions. We center the atoms in the unit cell and add vacuum on each side. We will add 6 Å of vacuum on each side. In the write command we use the option show_unit_cell =2 to draw the unit cell boundaries. See Figure ref:fig:ch3cn.
from ase.build import molecule
from ase.io import write
atoms = molecule('CH3CN')
atoms.center(vacuum=6)
print('unit cell')
print('---------')
print(atoms.get_cell())
write('images/ch3cn.png', atoms, show_unit_cell=2)
unit cell
---------
Cell([13.775328, 13.537479, 15.014576])
from ase.build import molecule
from ase.io import write
atoms = molecule('CH3CN')
atoms.center(vacuum=6)
print('unit cell')
print('---------')
print(atoms.get_cell())
write('images/ch3cn-rotated.png', atoms,
show_unit_cell=2, rotation='45x,45y,0z')
unit cell
---------
Cell([13.775328, 13.537479, 15.014576])
from ase.build import molecule
from ase.io import write
from numpy import pi
atoms = molecule('CH3CN')
atoms.center(vacuum=6)
p1 = atoms.get_positions()
atoms.rotate('x', pi/4, center='COM', rotate_cell=False)
atoms.rotate('y', pi/4, center='COM', rotate_cell=False)
write('images/ch3cn-rotated-2.png', atoms, show_unit_cell=2)
print('difference in positions after rotating')
print('atom difference vector')
print('--------------------------------------')
p2 = atoms.get_positions()
diff = p2 - p1
for i, d in enumerate(diff):
print('{0} {1}'.format(i, d))
difference in positions after rotating
atom difference vector
--------------------------------------
0 [-0.01782051 0.01782219 0.0002443 ]
1 [ 2.20136479e-03 -2.20157163e-03 -3.01777230e-05]
2 [ 0.01835166 -0.01835339 -0.00025158]
3 [-0.02277373 0.02287218 0.01436336]
4 [-0.02314601 0.02301662 -0.01887695]
5 [-0.02297922 0.02301662 0.0054581 ]
Combining Atoms objects#
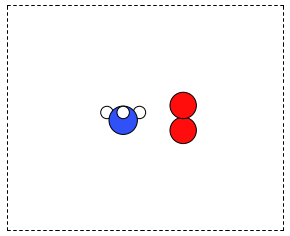
It is frequently useful to combine two Atoms objects, e.g. for computing reaction barriers, or other types of interactions. In ase, we simply add two Atoms objects together. Here is an example of getting an ammonia and oxygen molecule in the same unit cell. See Figure ref:fig:combined-atoms. We set the Atoms about three Å apart using the func:ase.Atoms.translate function.
from ase.build import molecule
from ase.io import write
atoms1 = molecule('NH3')
atoms2 = molecule('O2')
atoms2.translate([3, 0, 0])
bothatoms = atoms1 + atoms2
bothatoms.center(5)
write('images/bothatoms.png', bothatoms, show_unit_cell=2, rotation='90x')