A better manager for supervising Python functions
Posted September 21, 2023 at 01:42 PM | categories: programming | tags:
Updated September 21, 2023 at 01:43 PM
Table of Contents
In the previous post I introduced a supervisor decorator to automate rerunning functions with new arguments to fix issues in them. Almost immediately after posting it, two things started bugging me. First, I thought it was annoying to have two separate arguments for results and exceptions. I would prefer one list of functions that do the right thing. Second, and most annoying, you have to be very careful in writing your checker functions to be consistent with how you called the function so you use exactly the same positional and keyword arguments. That is tedious and limits reusability/flexibility.
So, I wrote a new manager decorator that solves these two problems. Now, you can write checker functions that work on all the arguments of a function. You decorate the checker functions to indicate if they are for results or exceptions. This was a little more of a rabbit hole than I anticipated, but I persevered, and got to a solution that works for these examples. You can find all the code here.
Here is an example where we have a test function that we want to run with new arguments until we get a positive result. We start in a way that it is possible to get a ZeroDivisionError, and we handle that too.
from pycse.supyrvisor import manager, check_result, check_exception @check_exception def check1(args, exc): if isinstance(exc, ZeroDivisionError): print('ooo. caught 1/0, incrementing x') return {'x': 1} @check_result def check2(args, result): print(args) if result < 0: args['x'] += 1 return args @manager(checkers=[check1, check2]) def test(x, a=1): return a / x test(-1)
{'x': -1, 'a': 1} ooo. caught 1/0, incrementing x {'x': 1} 1.0
This also works, so you can see this is better than the previous version which would not work if you change the signature.
test(a=1, x=-1)
{'x': -1, 'a': 1} ooo. caught 1/0, incrementing x {'x': 1} 1.0
1. The previous examples with manager
Here is the new syntax with manager.
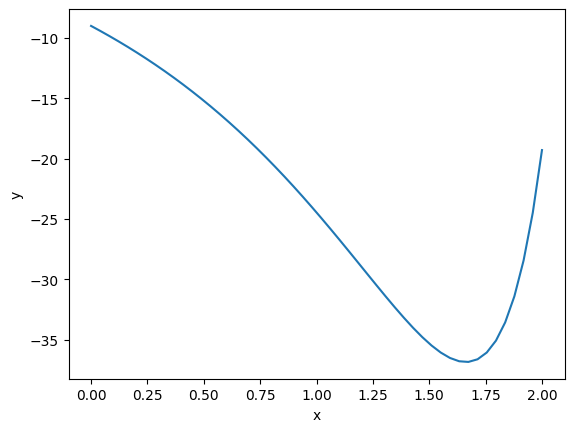
import numpy as np from scipy.optimize import minimize def objective(x): return np.exp(x**2) - 10 * np.exp(x) @check_result def maxIterationsExceeded(args, sol): if sol.message == 'Maximum number of iterations has been exceeded.': args['maxiter'] *= 2 return args @manager(checkers=[maxIterationsExceeded], verbose=True) def get_result(maxiter=2): return minimize(objective, 0.0, options={'maxiter': maxiter}) get_result(2)
Proposed fix in wrapper: {'maxiter': 4}
Proposed fix in wrapper: {'maxiter': 8}
message: Optimization terminated successfully.
success: True
status: 0
fun: -36.86307468296428
x: [ 1.662e+00]
nit: 5
jac: [-4.768e-07]
hess_inv: [[ 6.481e-03]]
nfev: 26
njev: 13
It works!
2. Stateful supervision
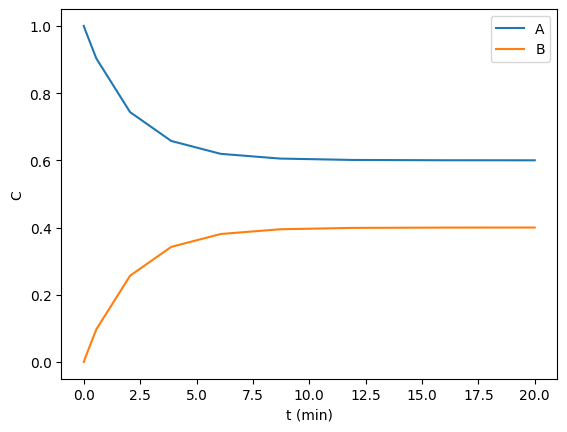
In this example, we aim to find the steady state concentrations of two species by integrating a mass balance to steady state. This is visually easy to see below, the concentrations are essentially flat after 10 min or so. Computationally this is somewhat tricky to find though. A way to do it is to compare some windows of integration to see if the values are not changing very fast. For instance you could average the values from 10 to 11, and compare that to the values in 11 to 12, and keep doing that until they are close enough to the same.
def ode(t, C): Ca, Cb = C dCadt = -0.2 * Ca + 0.3 * Cb dCbdt = -0.3 * Cb + 0.2 * Ca return dCadt, dCbdt tspan = (0, 20) from scipy.integrate import solve_ivp sol = solve_ivp(ode, tspan, (1, 0)) import matplotlib.pyplot as plt plt.plot(sol.t, sol.y.T) plt.xlabel('t (min)') plt.ylabel('C') plt.legend(['A', 'B']); sol.y.T[-1]
array([0.60003278, 0.39996722])
It is not crucial to use a class here; you could also use global variables, or function attributes. A class is a standard way of encapsulating state though. We just have to make the class callable so it acts like a function when we need it to.
class ReachedSteadyState: def __init__(self, tolerance=0.01): self.tolerance = tolerance self.last_solution = None self.count = 0 def __str__(self): return 'ReachedSteadyState' @check_result def __call__(self, args, sol): if self.last_solution is None: self.last_solution = sol self.count += 1 args['C0'] = sol.y.T[-1] return args # we have a previous solution if not np.allclose(self.last_solution.y.mean(axis=1), sol.y.mean(axis=1), rtol=self.tolerance, atol=self.tolerance): self.last_solution = sol self.count += 1 args['C0'] = sol.y.T[-1] return args rss = ReachedSteadyState(0.0001) @manager(checkers=[rss], max_errors=20, verbose=True) def get_sol(C0=(1, 0), window=1): sol = solve_ivp(ode, t_span=(0, window), y0=C0) return sol sol = get_sol((1, 0), window=2) sol
Proposed fix in ReachedSteadyState: {'C0': array([0.74716948, 0.25283052]), 'window': 2} Proposed fix in ReachedSteadyState: {'C0': array([0.65414484, 0.34585516]), 'window': 2} Proposed fix in ReachedSteadyState: {'C0': array([0.61992776, 0.38007224]), 'window': 2} Proposed fix in ReachedSteadyState: {'C0': array([0.60733496, 0.39266504]), 'window': 2} Proposed fix in ReachedSteadyState: {'C0': array([0.60269957, 0.39730043]), 'window': 2} Proposed fix in ReachedSteadyState: {'C0': array([0.60099346, 0.39900654]), 'window': 2} Proposed fix in ReachedSteadyState: {'C0': array([0.60036557, 0.39963443]), 'window': 2} Proposed fix in ReachedSteadyState: {'C0': array([0.60013451, 0.39986549]), 'window': 2} Proposed fix in ReachedSteadyState: {'C0': array([0.60004949, 0.39995051]), 'window': 2}
message: The solver successfully reached the end of the integration interval.
success: True
status: 0
t: [ 0.000e+00 7.179e-01 2.000e+00]
y: [[ 6.000e-01 6.000e-01 6.000e-01]
[ 4.000e-01 4.000e-01 4.000e-01]]
sol: None
t_events: None
y_events: None
nfev: 14
njev: 0
nlu: 0
We can plot the two solutions to see how different they are. This shows they are close.
import matplotlib.pyplot as plt plt.plot(rss.last_solution.t, rss.last_solution.y.T, label=['A previous' ,'B previous']) plt.plot(sol.t, sol.y.T, '--', label=['A current', 'B current']) plt.legend() plt.xlabel('relative t') plt.ylabel('C');
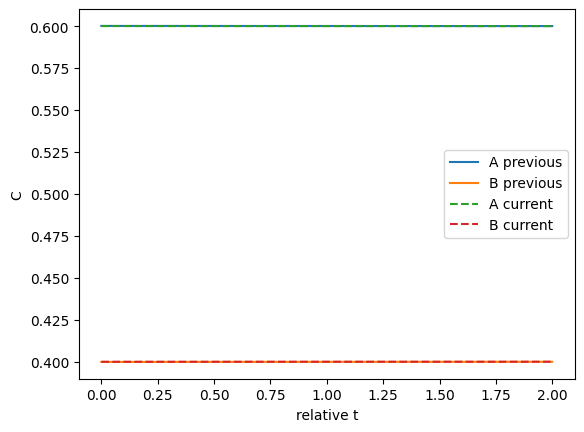
Those look pretty similar on this graph.
3. Handling exceptions
Suppose you have a function that randomly fails. This could be because something does not converge with a randomly chosen initial guess, converges to an unphysical answer, etc. In these cases, it makes sense to simply try again with a new initial guess.
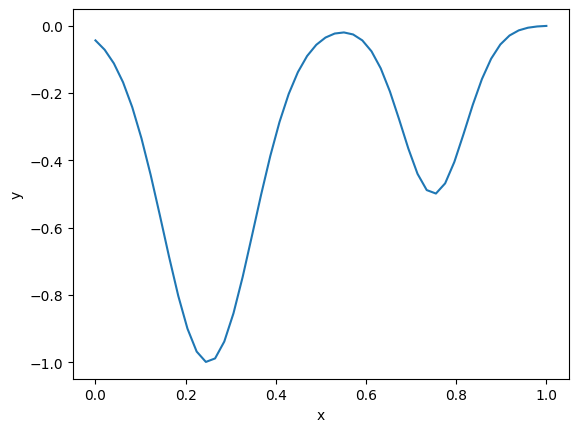
For this example, say we have this objective function with two minima. We will say that any solution above 0.5 is unphysical.
def f(x): return -(np.exp(-50 * (x - 0.25)**2) + 0.5 * np.exp(-100 * (x - 0.75)**2)) x = np.linspace(0, 1) plt.plot(x, f(x)) plt.xlabel('x') plt.ylabel('y');
Here we define a function that takes a guess, and gets a solution. If the solution is unphysical, we raise an exception. We define a custom exception so we can handle it specifically.
class UnphysicalSolution(Exception): pass def get_minima(guess): sol = minimize(f, guess) if sol.x > 0.5: raise UnphysicalSolution else: return sol @check_exception def try_again(args, exc): if isinstance(exc, UnphysicalSolution): args['guess'] = np.random.random() return args @manager(checkers=(try_again,), verbose=True) def get_minima(guess): sol = minimize(f, guess) if sol.x > 0.5: raise UnphysicalSolution else: return sol get_minima(np.random.random())
Proposed fix in wrapper: {'guess': 0.03789731690063758}
message: Optimization terminated successfully.
success: True
status: 0
fun: -1.0000000000069411
x: [ 2.500e-01]
nit: 4
jac: [ 0.000e+00]
hess_inv: [[ 1.000e-02]]
nfev: 18
njev: 9
You can see it took four iterations to find a solution. Other times it might take zero or one, or maybe more, it depends on where the guesses fall.
4. Summary
This solution works as well as supervisor did. It was a little deeper rabbit hole to go down, mostly because of some subtlety in making the result and exception decorators work for both functions and class methods. I think it is more robust now, as it should not matter how you call the function, and any combination of args and kwargs should be working.
Copyright (C) 2023 by John Kitchin. See the License for information about copying.
Org-mode version = 9.7-pre