06 - Band Structure#
This script calculates and plots the electronic band structure of siliconalong high-symmetry paths in the Brillouin zone.
import numpy as np
from ase.build import bulk
from vasp import Vasp
# Try to import matplotlib for plotting
try:
import matplotlib.pyplot as plt
HAS_MATPLOTLIB = True
except ImportError:
HAS_MATPLOTLIB = False
print("Note: matplotlib not found. Plots will be skipped.")
Step 1: Self-consistent calculation#
print("=" * 60)
print("Band Structure Calculation")
print("=" * 60)
print()
atoms = bulk('Si', 'diamond', a=5.43)
print("Step 1: Self-consistent ground state calculation")
print()
calc_scf = Vasp(
label='results/bands/scf',
atoms=atoms,
xc='PBE',
encut=400,
kpts=(8, 8, 8),
ismear=1,
sigma=0.1,
lwave=True,
lcharg=True,
)
energy = calc_scf.potential_energy
print(f" Total energy: {energy:.6f} eV")
print()
============================================================
Band Structure Calculation
============================================================
Step 1: Self-consistent ground state calculation
Total energy: -10.844419 eV
Step 2: Band structure calculation (non-self-consistent)#
print("Step 2: Non-self-consistent band structure calculation")
print()
# Define high-symmetry path for FCC Brillouin zone
# L-Gamma-X-U|K-Gamma
# In fractional coordinates of reciprocal lattice:
# L = (0.5, 0.5, 0.5)
# Gamma = (0, 0, 0)
# X = (0.5, 0, 0.5)
# U = (0.625, 0.25, 0.625)
# K = (0.375, 0.375, 0.75)
special_points = {
'L': [0.5, 0.5, 0.5],
'G': [0.0, 0.0, 0.0], # Gamma
'X': [0.5, 0.0, 0.5],
'U': [0.625, 0.25, 0.625],
'K': [0.375, 0.375, 0.75],
}
path = ['L', 'G', 'X', 'U', 'K', 'G']
# Generate k-points along the path
def generate_kpath(special_points, path, npoints_per_segment=20):
"""Generate k-points along high-symmetry path."""
kpts = []
labels = []
label_positions = []
for i, label in enumerate(path):
if i == 0:
labels.append(label)
label_positions.append(0)
elif label == path[i-1]:
# Skip duplicate (for discontinuous paths)
continue
else:
labels.append(label)
if i < len(path) - 1:
start = np.array(special_points[path[i]])
end = np.array(special_points[path[i+1]])
segment = np.linspace(start, end, npoints_per_segment, endpoint=False)
kpts.extend(segment.tolist())
if i == len(path) - 2:
kpts.append(end.tolist())
label_positions.append(len(kpts) - 1)
else:
label_positions.append(len(kpts))
return np.array(kpts), labels, label_positions
kpath, labels, label_positions = generate_kpath(special_points, path, npoints_per_segment=30)
print(f" Path: {' -> '.join(path)}")
print(f" Number of k-points: {len(kpath)}")
print()
calc_bands = Vasp(
label='results/bands/bands',
atoms=atoms,
xc='PBE',
encut=400,
kpts=kpath,
reciprocal=True, # K-points in fractional coordinates
ismear=0, # Gaussian smearing for bands
sigma=0.05,
icharg=11, # Read charge from SCF
lorbit=11, # For orbital character
lwave=False,
lcharg=False,
)
# Copy charge density from SCF calculation
import os
import shutil
scf_dir = calc_scf.directory
bands_dir = calc_bands.directory
os.makedirs(bands_dir, exist_ok=True)
for f in ['CHGCAR', 'CHG']:
src = os.path.join(scf_dir, f)
dst = os.path.join(bands_dir, f)
if os.path.exists(src):
shutil.copy(src, dst)
# Run calculation
_ = calc_bands.potential_energy
print(" Band calculation complete")
print()
Step 2: Non-self-consistent band structure calculation
Path: L -> G -> X -> U -> K -> G
Number of k-points: 151
Band calculation complete
Step 3: Read and analyze bands#
print("Step 3: Reading band data")
print()
# read_procar returns energies[nkpts, nbands] from PROCAR file
band_data = calc_bands.read_procar()
eigenvalues = band_data['energies']
fermi = calc_bands.results.get('efermi', 0.0)
print(f" Number of bands: {eigenvalues.shape[1]}")
print(f" Fermi level: {fermi:.4f} eV")
print()
# Calculate band gap
occupied = eigenvalues[eigenvalues < fermi]
unoccupied = eigenvalues[eigenvalues > fermi]
if len(occupied) > 0 and len(unoccupied) > 0:
vbm = occupied.max()
cbm = unoccupied.min()
band_gap = cbm - vbm
print(f" Valence band maximum: {vbm:.4f} eV")
print(f" Conduction band minimum: {cbm:.4f} eV")
print(f" Band gap: {band_gap:.3f} eV")
print(" Experimental band gap: 1.12 eV")
print()
Step 3: Reading band data
Number of bands: 8
Fermi level: 0.0000 eV
Valence band maximum: -0.0578 eV
Conduction band minimum: 0.0007 eV
Band gap: 0.059 eV
Experimental band gap: 1.12 eV
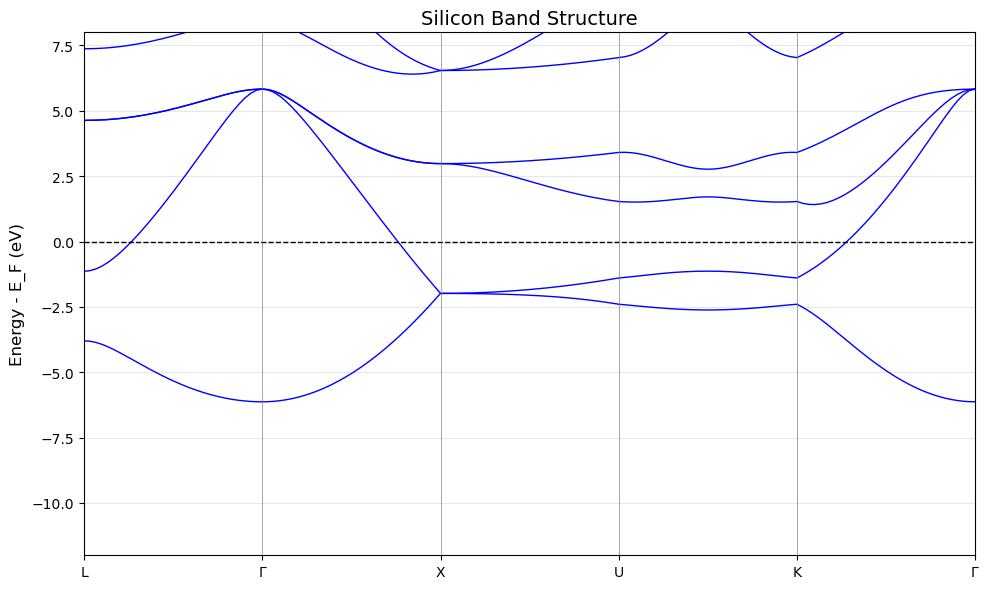
Step 4: Plot band structure#
if HAS_MATPLOTLIB:
print("Step 4: Plotting band structure")
print()
fig, ax = plt.subplots(figsize=(10, 6))
# Shift bands relative to Fermi level
bands_shifted = eigenvalues - fermi
# Plot each band
x = np.arange(len(kpath))
for i in range(bands_shifted.shape[1]):
ax.plot(x, bands_shifted[:, i], 'b-', linewidth=1)
# Add Fermi level
ax.axhline(y=0, color='k', linestyle='--', linewidth=1, label='results/Fermi level')
# Add vertical lines at high-symmetry points
for pos in label_positions:
ax.axvline(x=pos, color='gray', linestyle='-', linewidth=0.5)
# Labels
label_names = [r'$\Gamma$' if l == 'G' else l for l in labels]
ax.set_xticks(label_positions)
ax.set_xticklabels(label_names)
ax.set_ylabel('Energy - E_F (eV)', fontsize=12)
ax.set_title('Silicon Band Structure', fontsize=14)
ax.set_xlim(0, len(kpath) - 1)
ax.set_ylim(-12, 8)
ax.grid(True, axis='y', alpha=0.3)
plt.tight_layout()
plt.savefig('silicon_bands.png', dpi=150)
print(" Saved plot: silicon_bands.png")
print()
print("=" * 60)
print("Band structure calculation complete!")
print("=" * 60)
print()
print("Key points:")
print(" - Use non-self-consistent calculation (ICHARG=11)")
print(" - Define k-path through high-symmetry points")
print(" - Silicon is an indirect gap semiconductor")
print(" - VBM at Gamma, CBM near X")
print()
print("Next: Try 07_magnetism/ for spin-polarized calculations.")
Step 4: Plotting band structure
Saved plot: silicon_bands.png
============================================================
Band structure calculation complete!
============================================================
Key points:
- Use non-self-consistent calculation (ICHARG=11)
- Define k-path through high-symmetry points
- Silicon is an indirect gap semiconductor
- VBM at Gamma, CBM near X
Next: Try 07_magnetism/ for spin-polarized calculations.