First post on DFT book blog
We have a new rewrite of the vasp Python calculator (https://github.com/jkitchin/vasp). Most of DFT-book has been re-written with the new library to provide testing and examples. There are a lot of new ideas in it though, so I am creating this new blog for dft-book to try them out, discuss various design choices, and develop advanced methods in running DFT calculations. This is the first post!
One of the features it has is better support for things like list comprehensions in DFT calculations. Here is an example of that where we start with a list of bond lengths, create a list of calculators, and get the energy from each one. This one script sets up and runs the calculations. You can run it over and over, and it should simply retrieve the results once the calculations are finished.
from vasp import Vasp from ase import Atom, Atoms import matplotlib.pyplot as plt bond_lengths = [1.05, 1.1, 1.15, 1.2, 1.25] calcs = [Vasp('molecules/co-{0}'.format(d), # output dir xc='PBE', nbands=6, encut=350, ismear=1, sigma=0.01, atoms=Atoms([Atom('C', [0, 0, 0]), Atom('O', [d, 0, 0])], cell=(6, 6, 6))) for d in bond_lengths] energies = [calc.potential_energy for calc in calcs] # Stop here until all calculations are done. calcs[0].stop_if(None in energies) plt.plot(energies) plt.xlabel('Bond length ($\AA$)') plt.ylabel('Energy (eV)') plt.savefig('test.png')
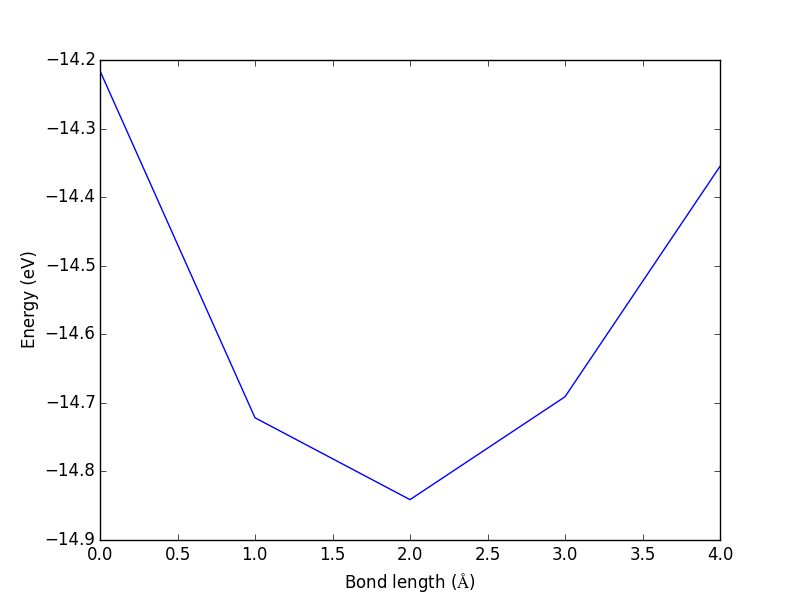
Figure 1: Energy vs. bond-length for the CO molecule.
Comments
Comments powered by Disqus