Posted September 24, 2023 at 03:04 PM | categories:
news, publication | tags:
In this work we show that prediction errors from graph neural networks for related atomistic systems tend to be correlated, and as a result the differences in energy are more accurate than the absolute energies. This is similar to what is observed in DFT calculations where systematic errors also cancel in differences. We show this quantitatively through differences of systems with systematically different levels of similarity. This article is part of the Special Collection: 2023 JCP Emerging Investigators Special Collection.
@article{ock-2023-beyon-indep,
author = {Janghoon Ock and Tian Tian and John Kitchin and Zachary
Ulissi},
title = {Beyond Independent Error Assumptions in Large {GNN} Atomistic
Models},
journal = {The Journal of Chemical Physics},
volume = 158,
number = 21,
pages = {nil},
year = 2023,
doi = {10.1063/5.0151159},
url = {http://dx.doi.org/10.1063/5.0151159},
DATE_ADDED = {Sun Sep 24 14:53:46 2023},
}
Copyright (C) 2023 by John Kitchin. See the License for information about copying.
I have been working on hashcache to make it more flexible. I like the base functionality that uses the filesystem for caching. That still works.
Here I set up a timeit decorator to show how this works.
from pycse.hashcache import hashcache
import time
!rm -fr ./cache
deftimeit(func):
defwrapper(*args, **kwargs):
t0 = time.time()
res = func(*args, **kwargs)
print(f'Elapsed time = {time.time() - t0}s')
return res
return wrapper
Now we decorate a function that is "expensive". The first time we run it, it takes a long time.
@timeit@hashcachedefexpensive_func(x):
time.sleep(3)
return x
expensive_func(2)
Elapsed time = 3.007030963897705s
2
The second time is very fast, since we just look it up.
expensive_func(2)
Elapsed time = 0.0012097358703613281s
2
Where did we look it up from? It is stored on disk. You can see where by adding a verbose option to the decorator. This shows you all the data that was stored in the cache.
@hashcache(verbose=True)
defexpensive_func(x):
time.sleep(3)
return x
expensive_func(2)
The file system is an amazing cache with many benefits. There are few reasons you might like something different though. For example, it is slow to search if you have to iterate over all the directories and read the files, and it might be slow to sync lots of directories to another place.
hashcache is more flexible now, so you can define the functions that load and dump the cache. Here we use lmdb as a key-value database. lmdb expects the keys and values to be bytes, so we do some tricks with io.BytesIO to get these as strings from joblib.dump which expects to write to a file.
The load function has the signature (hash, verbose), and the dump function has the signature (hash, data, verbose). In both cases, hash will be a string for the key to save data in. data will be a dictionary that should be saved in a way that it can be reloaded. verbose is a flag that you can ignore or use to provide some kind of logging.
from pycse.hashcache import hashcache
import io, joblib, lmdb
deflmdb_dump(hsh, data, verbose=False):
if verbose:
print('running lmdb_dump')
with io.BytesIO() as f:
joblib.dump(data, f)
value = f.getvalue()
db = lmdb.Environment(hashcache.cache)
with db.begin(write=True) as txn:
txn.put(hsh.encode('utf-8'), value)
deflmdb_load(hsh, verbose=False):
if verbose:
print('running lmdb_load')
db = lmdb.Environment(hashcache.cache)
with db.begin() as txn:
val = txn.get(hsh.encode('utf-8'))
if val isNone:
returnFalse, Noneelse:
returnTrue, joblib.load(io.BytesIO(val))['output']
! rm -fr cache.lmdb
hashcache.cache = 'cache.lmdb'@hashcache(loader=lmdb_load, dumper=lmdb_dump, verbose=True)
deff(x):
return x
f(2)
running lmdb_load
running lmdb_dump
2
And we can recall the result as easily.
f(2)
running lmdb_load
2
2. a shelve version
Maybe you prefer a built in library like shelve. This is also quite simple.
from pycse.hashcache import hashcache
import io, joblib, shelve
defshlv_dump(hsh, data, verbose=False):
print('running shlv_dump')
with io.BytesIO() as f:
joblib.dump(data, f)
value = f.getvalue()
with shelve.open(hashcache.cache) as db:
db[hsh] = value
defshlv_load(hsh, verbose=False):
print('running shlv_load')
with shelve.open(hashcache.cache) as db:
if hsh in db:
returnTrue, joblib.load(io.BytesIO(db[hsh]))['output']
else:
returnFalse, None
hashcache.cache = 'cache.shlv'
! rm -f cache.shlv.db
@hashcache(loader=shlv_load, dumper=shlv_dump)
deff(x):
return x
f(2)
running shlv_load
running shlv_dump
2
And again loading is easy.
f(2)
running shlv_load
2
3. sqlite version
I am a big fan of sqlite. Here I use a simple table mapping a key to a value. I think it could be interesting to consider storing the value as json that would make it more searchable, or you could make a more complex table, but here we keep it simple.
from pycse.hashcache import hashcache
import io, joblib, sqlite3
defsql_dump(hsh, data, verbose=False):
print('running sql_dump')
with io.BytesIO() as f:
joblib.dump(data, f)
value = f.getvalue()
with con:
con.execute("INSERT INTO cache(hash, value) VALUES(?, ?)",
(hsh, value))
defsql_load(hsh, verbose=False):
print('running sql_load')
with con:
cur = con.execute("SELECT value FROM cache WHERE hash = ?",
(hsh,))
value = cur.fetchone()
if value isNone:
returnFalse, Noneelse:
returnTrue, joblib.load(io.BytesIO(value[0]))['output']
! rm -f cache.sql
hashcache.cache = 'cache.sql'
con = sqlite3.connect(hashcache.cache)
con.execute("CREATE TABLE cache(hash TEXT unique, value BLOB)")
@hashcache(loader=sql_load, dumper=sql_dump)
deff(x):
return x
f(2)
running sql_load
running sql_dump
2
Once again, running is easy.
f(2)
running sql_load
2
4. redis
Finally, you might like a server to cache in. This opens the door to running the server remotely so it is accessible by multiple processes using the cache on different machines. We use redis for this example, but only run it locally. Make sure you run redis-server --daemonize yes
from pycse.hashcache import hashcache
import io, joblib, redis
db = redis.Redis(host='localhost', port=6379)
defredis_dump(hsh, data, verbose=False):
print('running redis_dump')
with io.BytesIO() as f:
joblib.dump(data, f)
value = f.getvalue()
db.set(hsh, value)
defredis_load(hsh, verbose=False):
print('running redis_load')
ifnot hsh in db:
returnFalse, Noneelse:
returnTrue, joblib.load(io.BytesIO(db.get(hsh)))['output']
import functools
hashcache_redis = functools.partial(hashcache,
loader=redis_load,
dumper=redis_dump)
@hashcache_redisdeff(x):
return x
f(2)
running redis_load
running redis_dump
2
No surprise here, loading is the same as before.
f(2)
running redis_load
2
5. Summary
I have refactored hashcache to make it much easier to add new backends. You might do that for performance, ease of backup or transferability, to add new capabilities for searching, etc. The new code is a little cleaner than it was before IMO. I am not sure it is API-stable yet, but it is getting there.
Copyright (C) 2023 by John Kitchin. See the License for information about copying.
In the previous post I introduced a supervisor decorator to automate rerunning functions with new arguments to fix issues in them. Almost immediately after posting it, two things started bugging me. First, I thought it was annoying to have two separate arguments for results and exceptions. I would prefer one list of functions that do the right thing. Second, and most annoying, you have to be very careful in writing your checker functions to be consistent with how you called the function so you use exactly the same positional and keyword arguments. That is tedious and limits reusability/flexibility.
So, I wrote a new manager decorator that solves these two problems. Now, you can write checker functions that work on all the arguments of a function. You decorate the checker functions to indicate if they are for results or exceptions. This was a little more of a rabbit hole than I anticipated, but I persevered, and got to a solution that works for these examples. You can find all the code here.
Here is an example where we have a test function that we want to run with new arguments until we get a positive result. We start in a way that it is possible to get a ZeroDivisionError, and we handle that too.
from pycse.supyrvisor import manager, check_result, check_exception
@check_exceptiondefcheck1(args, exc):
ifisinstance(exc, ZeroDivisionError):
print('ooo. caught 1/0, incrementing x')
return {'x': 1}
@check_resultdefcheck2(args, result):
print(args)
if result < 0:
args['x'] += 1
return args
@manager(checkers=[check1, check2])
deftest(x, a=1):
return a / x
test(-1)
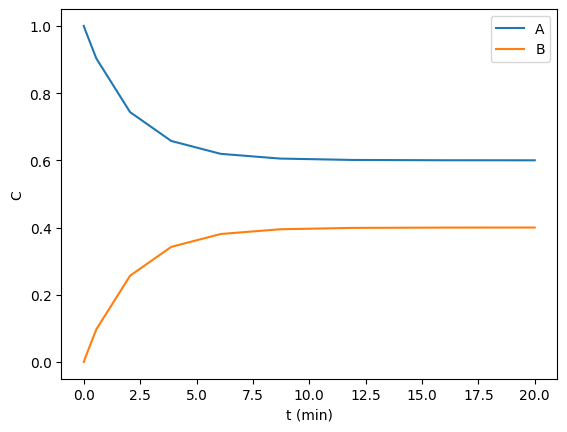
In this example, we aim to find the steady state concentrations of two species by integrating a mass balance to steady state. This is visually easy to see below, the concentrations are essentially flat after 10 min or so. Computationally this is somewhat tricky to find though. A way to do it is to compare some windows of integration to see if the values are not changing very fast. For instance you could average the values from 10 to 11, and compare that to the values in 11 to 12, and keep doing that until they are close enough to the same.
defode(t, C):
Ca, Cb = C
dCadt = -0.2 * Ca + 0.3 * Cb
dCbdt = -0.3 * Cb + 0.2 * Ca
return dCadt, dCbdt
tspan = (0, 20)
from scipy.integrate import solve_ivp
sol = solve_ivp(ode, tspan, (1, 0))
import matplotlib.pyplot as plt
plt.plot(sol.t, sol.y.T)
plt.xlabel('t (min)')
plt.ylabel('C')
plt.legend(['A', 'B']);
sol.y.T[-1]
array([0.60003278, 0.39996722])
It is not crucial to use a class here; you could also use global variables, or function attributes. A class is a standard way of encapsulating state though. We just have to make the class callable so it acts like a function when we need it to.
classReachedSteadyState:
def__init__(self, tolerance=0.01):
self.tolerance = tolerance
self.last_solution = Noneself.count = 0
def__str__(self):
return'ReachedSteadyState'@check_resultdef__call__(self, args, sol):
ifself.last_solution isNone:
self.last_solution = sol
self.count += 1
args['C0'] = sol.y.T[-1]
return args
# we have a previous solutionifnot np.allclose(self.last_solution.y.mean(axis=1),
sol.y.mean(axis=1),
rtol=self.tolerance,
atol=self.tolerance):
self.last_solution = sol
self.count += 1
args['C0'] = sol.y.T[-1]
return args
rss = ReachedSteadyState(0.0001)
@manager(checkers=[rss], max_errors=20, verbose=True)
defget_sol(C0=(1, 0), window=1):
sol = solve_ivp(ode, t_span=(0, window), y0=C0)
return sol
sol = get_sol((1, 0), window=2)
sol
Proposed fix in ReachedSteadyState: {'C0': array([0.74716948, 0.25283052]), 'window': 2}
Proposed fix in ReachedSteadyState: {'C0': array([0.65414484, 0.34585516]), 'window': 2}
Proposed fix in ReachedSteadyState: {'C0': array([0.61992776, 0.38007224]), 'window': 2}
Proposed fix in ReachedSteadyState: {'C0': array([0.60733496, 0.39266504]), 'window': 2}
Proposed fix in ReachedSteadyState: {'C0': array([0.60269957, 0.39730043]), 'window': 2}
Proposed fix in ReachedSteadyState: {'C0': array([0.60099346, 0.39900654]), 'window': 2}
Proposed fix in ReachedSteadyState: {'C0': array([0.60036557, 0.39963443]), 'window': 2}
Proposed fix in ReachedSteadyState: {'C0': array([0.60013451, 0.39986549]), 'window': 2}
Proposed fix in ReachedSteadyState: {'C0': array([0.60004949, 0.39995051]), 'window': 2}
message: The solver successfully reached the end of the integration interval.
success: True
status: 0
t: [ 0.000e+00 7.179e-01 2.000e+00]
y: [[ 6.000e-01 6.000e-01 6.000e-01]
[ 4.000e-01 4.000e-01 4.000e-01]]
sol: None
t_events: None
y_events: None
nfev: 14
njev: 0
nlu: 0
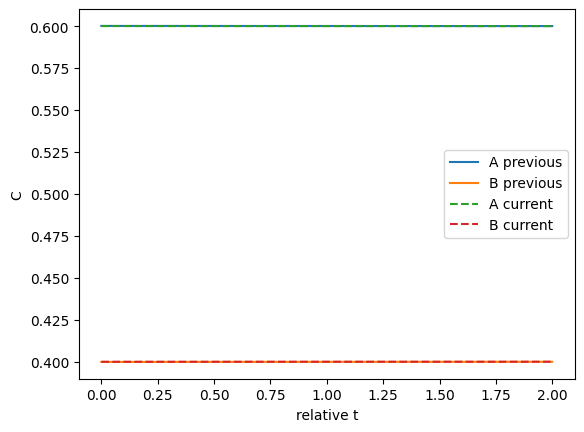
We can plot the two solutions to see how different they are. This shows they are close.
Suppose you have a function that randomly fails. This could be because something does not converge with a randomly chosen initial guess, converges to an unphysical answer, etc. In these cases, it makes sense to simply try again with a new initial guess.
For this example, say we have this objective function with two minima. We will say that any solution above 0.5 is unphysical.
Here we define a function that takes a guess, and gets a solution. If the solution is unphysical, we raise an exception. We define a custom exception so we can handle it specifically.
classUnphysicalSolution(Exception):
passdefget_minima(guess):
sol = minimize(f, guess)
if sol.x > 0.5:
raise UnphysicalSolution
else:
return sol
@check_exceptiondeftry_again(args, exc):
ifisinstance(exc, UnphysicalSolution):
args['guess'] = np.random.random()
return args
@manager(checkers=(try_again,), verbose=True)
defget_minima(guess):
sol = minimize(f, guess)
if sol.x > 0.5:
raise UnphysicalSolution
else:
return sol
get_minima(np.random.random())
You can see it took four iterations to find a solution. Other times it might take zero or one, or maybe more, it depends on where the guesses fall.
4. Summary
This solution works as well as supervisor did. It was a little deeper rabbit hole to go down, mostly because of some subtlety in making the result and exception decorators work for both functions and class methods. I think it is more robust now, as it should not matter how you call the function, and any combination of args and kwargs should be working.
Copyright (C) 2023 by John Kitchin. See the License for information about copying.
[UPDATE [2023-09-21 Thu]]: See this new post for an update and improved version of this post.
In the last post I talked about using custodian to supervise Python functions. I noted it felt a little heavy, so I wrote a new decorator that does basically the same thing. TL;DR I am not sure this is less heavy, but I learned some things doing it. The code I used is part of pycse at https://github.com/jkitchin/pycse/blob/master/pycse/supyrvisor.py. Check out the code to see how this works.
Here is the prototype problem it solves. This code runs, but does not succeed because it exceeds the maximum iterations.
import numpy as np
from scipy.optimize import minimize
defobjective(x):
return np.exp(x**2) - 10 * np.exp(x)
minimize(objective, 0.0, options={'maxiter': 2})
message: Maximum number of iterations has been exceeded.
success: False
status: 1
fun: -36.86289091418059
x: [ 1.661e+00]
nit: 2
jac: [-2.374e-01]
hess_inv: [[ 6.889e-03]]
nfev: 20
njev: 10
The solution is simple, you increase the number of iterations. That is tedious to do manually though, and not practical if you do this hundreds of times in a study. Enter pycse.supyrvisor. It provides a decorator to do this. Similar to custodian, we have to define a function that has arguments to change this. We do this here. This function still does not succeed yet.
message: Maximum number of iterations has been exceeded.
success: False
status: 1
fun: -36.86289091418059
x: [ 1.661e+00]
nit: 2
jac: [-2.374e-01]
hess_inv: [[ 6.889e-03]]
nfev: 20
njev: 10
Next, we need a "checker" function. The role of this function is to check the output of the function, determine if it is ok, and if not, to return a new set of arguments to run the function with. There are some subtleties in this. You can call your function with a combination of args and kwargs, and you have to write this function in a way that is consistent with how you call the function. In the example above, we called get_result(2) where the 2 is a positional argument. In this checker function we write it with that in mind. If we detect that the minimizer failed because of exceeding the maximum number of iterations, we get the argument and double it. Then, we return the new args and kwargs. Otherwise this function returns None, indicating the solution is fine as far as this function is concerned.
defmaxIterationsExceeeded(args, kwargs, sol):
if sol.message == 'Maximum number of iterations has been exceeded.':
maxiter = args[0]
maxiter *= 2
return (maxiter,), kwargs
Finally, we get the supervisor decorator, and decorate the function.
from pycse.supyrvisor import supervisor
get_result = supervisor(check_funcs=[maxIterationsExceeeded], verbose=True)(get_result)
get_result(2)
In this example, we aim to find the steady state concentrations of two species by integrating a mass balance to steady state. This is visually easy to see below, the concentrations are essentially flat after 10 min or so. Computationally this is somewhat tricky to find though. A way to do it is to compare some windows of integration to see if the values are not changing very fast. For instance you could average the values from 10 to 11, and compare that to the values in 11 to 12, and keep doing that until they are close enough to the same.
defode(t, C):
Ca, Cb = C
dCadt = -0.2 * Ca + 0.3 * Cb
dCbdt = -0.3 * Cb + 0.2 * Ca
return dCadt, dCbdt
tspan = (0, 20)
from scipy.integrate import solve_ivp
sol = solve_ivp(ode, tspan, (1, 0))
import matplotlib.pyplot as plt
plt.plot(sol.t, sol.y.T)
plt.xlabel('t (min)')
plt.ylabel('C')
plt.legend(['A', 'B']);
sol.y.T[-1]
array([0.60003278, 0.39996722])
The goal then is to have a supervisor function that will keep track of the last solution and the current one, and compare the average of them. You could do something more sophisticated, but this is simple enough to try out now. If the difference between two integrations is small enough, we will say we have hit steady state, and if not, we integrate from the end of the last solution forward again. That means we have to store some state information so we can compare a current solution to the last solution.
Let's start by defining a function that returns a solution from some initial condition. Next, we show that if you run it 12ish times, initializing from the last state, we get something that appears steady-stateish in the sense that the y values only changing in the second decimal place. You might consider that close enough to steady state.
defget_sol(C0=(1, 0), window=1):
sol = solve_ivp(ode, t_span=(0, window), y0=C0)
return sol
sol = get_sol()
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol = get_sol(sol.y.T[-1])
sol
message: The solver successfully reached the end of the integration interval.
success: True
status: 0
t: [ 0.000e+00 3.565e-01 1.000e+00]
y: [[ 6.016e-01 6.014e-01 6.010e-01]
[ 3.984e-01 3.986e-01 3.990e-01]]
sol: None
t_events: None
y_events: None
nfev: 14
njev: 0
nlu: 0
That is obviously tedious, so now we devise a supervisor function to do it for us. Since we will save state between calls, I will use a class here. We will define a tolerance that we want the difference of the average of two sequential solutions to be less than. We have to be a little careful here. There are many ways to call get_sol, e.g. all of these are correct, but when the checker function is called, it will get different arguments.
get_sol() # no args: args=(), kwargs={}
get_sol((1, 0), 2) # all positional args: args=((1, 0), 2), kwargs={}
get_sol((1, 0)) # one positional arg: args=((1, 0),), kwargs={}
get_sol((1, 0), window=2) # a positional and kwarg: args =((1, 0),), kwargs={'window': 2}
We have to either assume one of these, or write a function that can handle any of them. I am going to assume here that args will always just be the initial condition, and anything else will be in kwargs. That is a convention we use for this problem, and if you break the convention, you will have errors. For example, get_sol(C0=(1, 0)) will cause an error because you will not have a positional argument for C0 but instead a keyword argument for C0.
It is not crucial to use a class here; you could also use global variables, or function attributes. A class is a standard way of encapsulating state though. We just have to make the class callable so it acts like a function when we need it to.
Now, we decorate the get_sol function, and then run it. Since we used a bigger window, it only takes 9 iterations to get to an approximate steady state.
defget_sol(C0=(1, 0), window=1):
sol = solve_ivp(ode, t_span=(0, window), y0=C0)
return sol
rss = ReachedSteadyState(0.0001)
get_sol = supervisor(check_funcs=(rss,), verbose=True, max_errors=20)(get_sol)
sol = get_sol((1, 0), window=2)
sol
Proposed fix in ReachedSteadyState: ((array([0.74716948, 0.25283052]),), {'window': 2})
Proposed fix in ReachedSteadyState: ((array([0.65414484, 0.34585516]),), {'window': 2})
Proposed fix in ReachedSteadyState: ((array([0.61992776, 0.38007224]),), {'window': 2})
Proposed fix in ReachedSteadyState: ((array([0.60733496, 0.39266504]),), {'window': 2})
Proposed fix in ReachedSteadyState: ((array([0.60269957, 0.39730043]),), {'window': 2})
Proposed fix in ReachedSteadyState: ((array([0.60099346, 0.39900654]),), {'window': 2})
Proposed fix in ReachedSteadyState: ((array([0.60036557, 0.39963443]),), {'window': 2})
Proposed fix in ReachedSteadyState: ((array([0.60013451, 0.39986549]),), {'window': 2})
Proposed fix in ReachedSteadyState: ((array([0.60004949, 0.39995051]),), {'window': 2})
message: The solver successfully reached the end of the integration interval.
success: True
status: 0
t: [ 0.000e+00 7.179e-01 2.000e+00]
y: [[ 6.000e-01 6.000e-01 6.000e-01]
[ 4.000e-01 4.000e-01 4.000e-01]]
sol: None
t_events: None
y_events: None
nfev: 14
njev: 0
nlu: 0
We can plot the two solutions to see how different they are. This shows they are close.
Suppose you have a function that randomly fails. This could be because something does not converge with a randomly chosen initial guess, converges to an unphysical answer, etc. In these cases, it makes sense to simply try again with a new initial guess.
For this example, say we have this objective function with two minima. We will say that any solution above 0.5 is unphysical.
Here we define a function that takes a guess, and gets a solution. If the solution is unphysical, we raise an exception. We define a custom exception so we can handle it specifically.
classUnphysicalSolution(Exception):
passdefget_minima(guess):
sol = minimize(f, guess)
if sol.x > 0.5:
raise UnphysicalSolution
else:
return sol
You can see it took two iterations to find a solution. Other times it might take zero or one, or maybe more, it depends on where the guesses fall.
3. Summary
This solution works pretty well, similar to custodian. It is a little simpler than custodian I think, as you can do simple things with functions, and don't really need to make classes for everything. Probably it does less than custodian, and also probably there are some corner issues I haven't uncovered yet. It was a nice exercise in building a decorator though, and thinking through all the ways this can be done.
Copyright (C) 2023 by John Kitchin. See the License for information about copying.
Posted September 19, 2023 at 03:57 PM | categories:
news, publication | tags:
We have a new publication out! In this work we show how to use classification algorithms to find boundaries in science and engineering applications. These applications come up all over the place, for example you may want to know what compositions phase separate, and which ones are single phase, or what conditions lead to degradation and which ones don't. You might want to know which operating parameters have desirable properties, and which don't. In this work we show how to efficiently find the boundaries between these regions using active learning and a classifier. We show that this approach is generally better (more accurate and fewer experiments) than doing a dense grid search.
@article{bhat-2023-sequen-sampl,
author = {Maya Bhat and John R. Kitchin},
title = {Sequential Sampling Methods for Finding Classification
Boundaries in Engineering Applications},
journal = {Industrial \& Engineering Chemistry Research},
volume = {n/a},
number = {n/a},
pages = {n/a},
year = 2023,
doi = {10.1021/acs.iecr.3c02362},
url = {http://dx.doi.org/10.1021/acs.iecr.3c02362}
}
Check out the Youtube video summary here:
Copyright (C) 2023 by John Kitchin. See the License for information about copying.