A little more than a decade of the Kitchingroup blog
Posted April 03, 2024 at 08:38 AM | categories: uncategorized | tags:
There are a few early entries I backdated, but this blog got started in its present form in January 2013. This entry marks entry #594. I started this blog as part of an exercise in switching from Matlab to Python, and the first hundred entries or so are just me solving a problem in Python that I had previously solved in Matlab. It then expanded to include lots of entries on Emacs and org-mode, and other research related topics from my group. Many entries simply document something I spent time working out and that I wanted to be able to find by Google later.
When I set the blog up, I enabled Google Analytics to see if anyone would look at. Recently Google announced they are shutting down the version of analytics I was using, and transitioning to a newer approach. They no longer collect data with the version this blog is using (since Oct last year), and they will delete the data this summer, so today I downloaded some of it to see what has happened over the past decade.
Anecdotally many people from around the world have told me how useful the blog was for them. Now, I have data to see how many people have been impacted by this blog. This figure shows that a lot of people spent time in some part of the blog over the past decade! The data suggests over 1M people viewed these pages over 2M times.
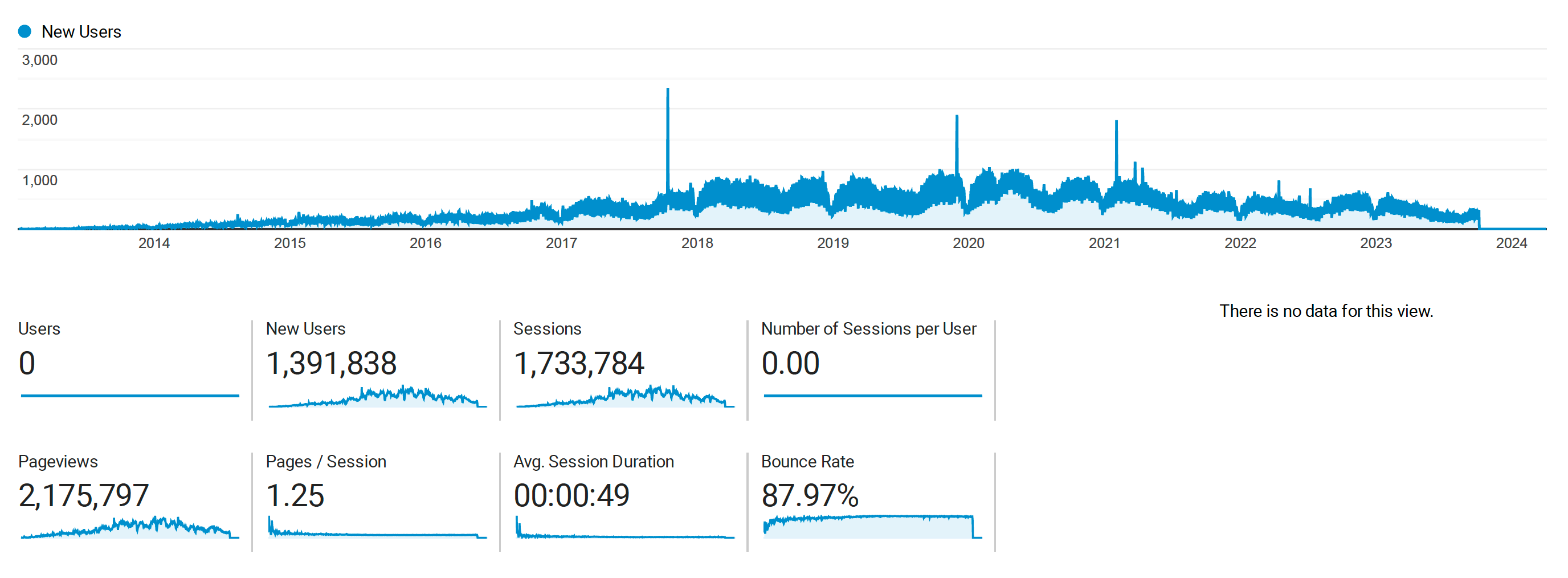
The peak usage was around 2020, and it has been trailing off since then. I have not been as active in posting since then. You can also see there is a very long build up to that peak.
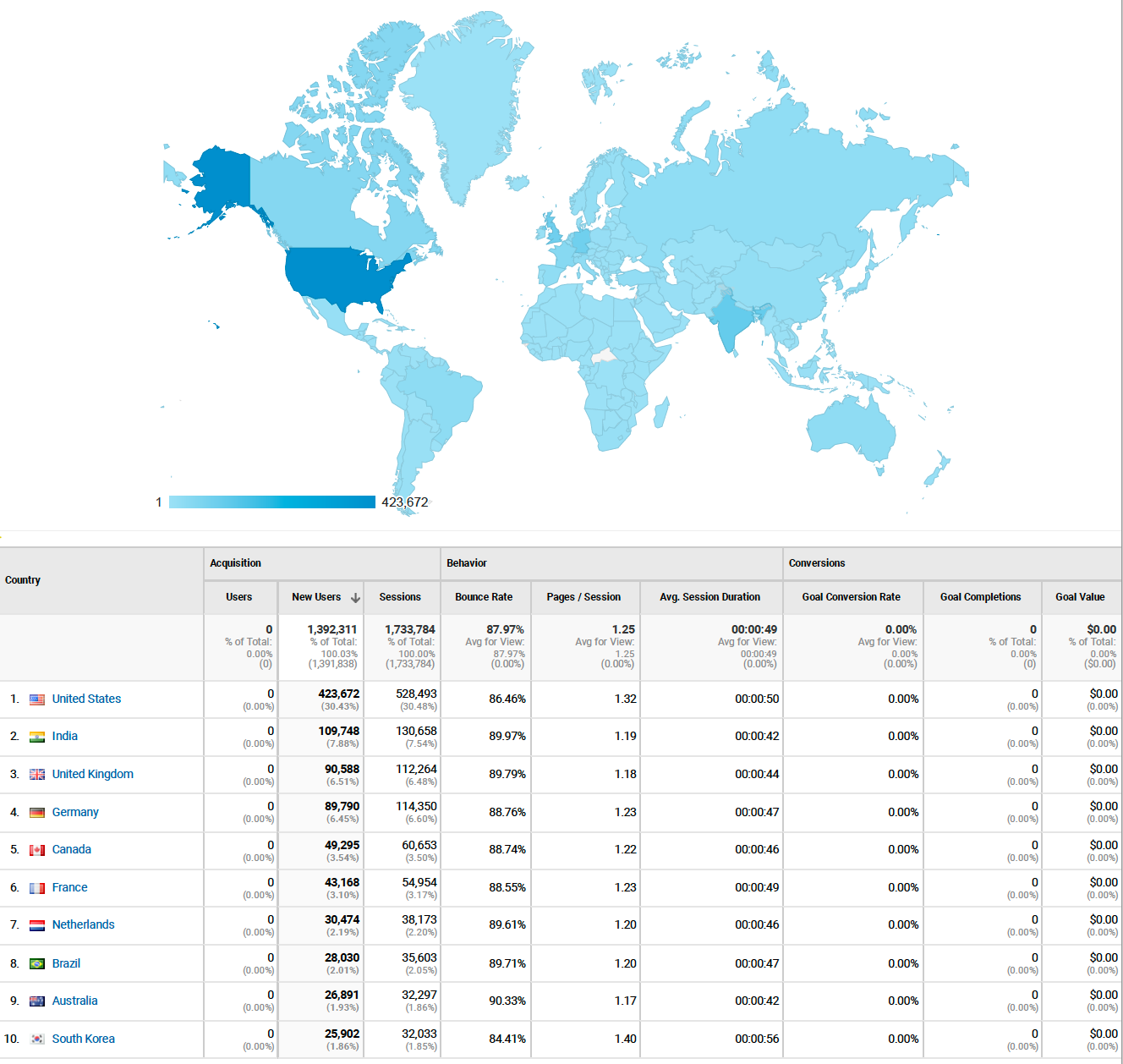
The user group for the blog is truly world wide, including almost every country in this map. That is amazing!
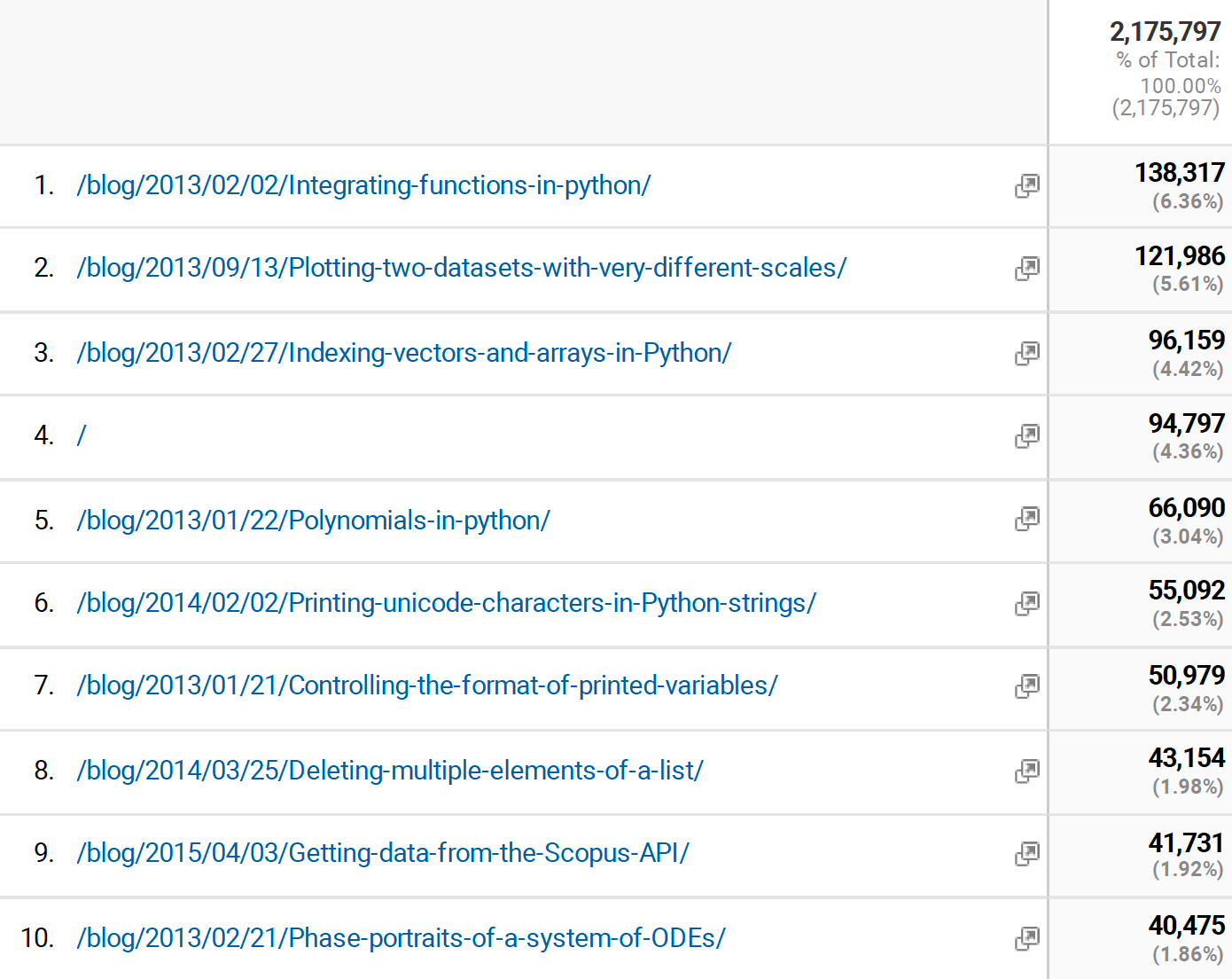
Finally, I found the pages that were most viewed. It is interesting most of them are the older pages, and all about Python. I guess that means I should write more posts on Python.
I don't know what the future of the blog is. It is in need of an overhaul. The packages that build it still work, but are not actively maintained. I have also spent more time writing with Jupyter Book lately than the way I wrote this blog. It isn't likely to disappear any time soon, it sits rent-free in GitHUB pages.
To conclude, to everyone who has read these pages, thank you! It has been a lot of work to put together over the years, and I am glad to see many people have taken a look at it.
Copyright (C) 2024 by John Kitchin. See the License for information about copying.
Org-mode version = 9.7-pre